
 商标分类
商标分类  商标转让
商标转让 
一种复合发光材料、其制备方法及其应用与流程
 2021-02-02 18:02:11|
2021-02-02 18:02:11| 398|
398| 起点商标网
起点商标网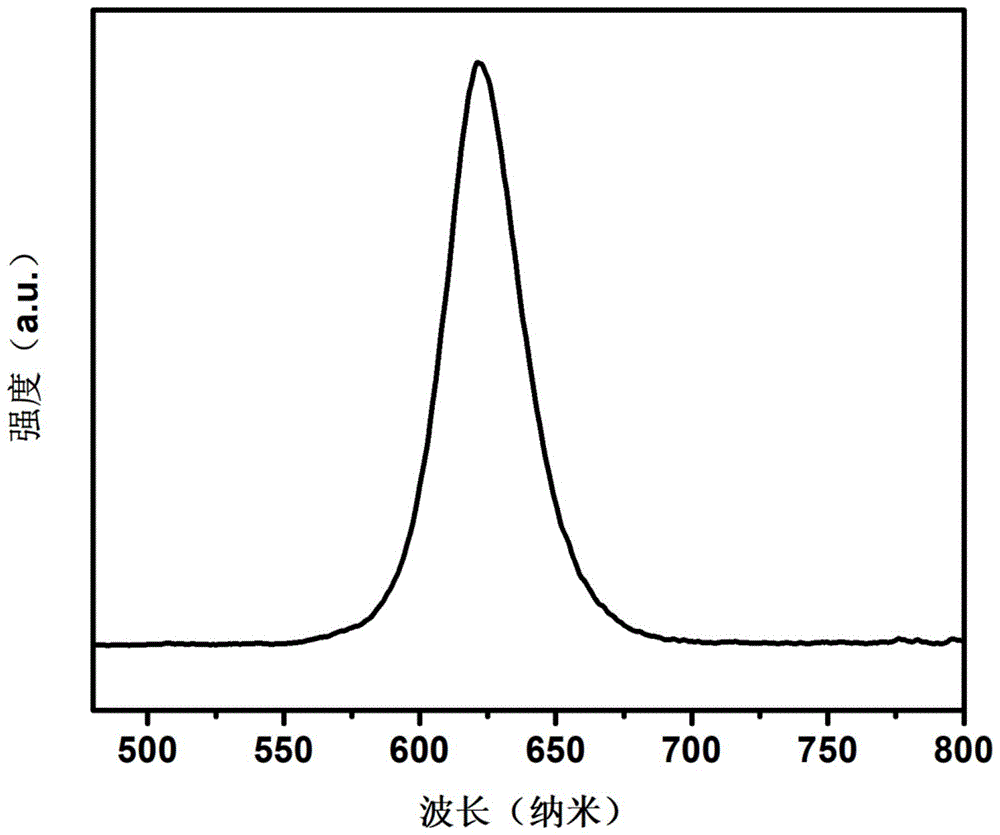
本申请涉及一种复合发光材料,属于材料领域。
背景技术:
钙钛矿材料的结构式一般为abx3,其中a可以是k+、na+、rb+、cs+以及小分子有机阳离子等,b可以是pb2+、sn2+、ti4+、cr3+、bi3+等多种元素,x可以由o2-、cl-、br-、i-、s2-等阴离子组成。钙钛矿结构的材料具备了很多独特的理化性质,比如吸光性、电催化性等等,在化学、物理领域有着广泛的应用。卤素钙钛矿是一类x为卤素(cl、br、i)阴离子的钙钛矿结构化合物,其中a可以是小分子有机阳离子,也可以是无机金属阳离子,分别称为有机-无机杂化卤素钙钛矿和全无机卤素钙钛矿。卤素钙钛矿的晶体结构由1个b金属原子和6个x原子构成1个八面体结构,a原子镶嵌在8个八面体两两共用1个x原子形成的立方体结构的中心。卤素钙钛矿材料具有独特的光电半导体特性,其具有合适的带隙、较高的载流子迁移率、很强的缺陷容忍性、较低的浅点缺陷率、较低的晶界复合率和表面复合率,以及由于s-p反键耦合产生的较大的光吸收系数。这些独特的光学和半导体特性使得卤素钙钛矿材料成为太阳能电池中光电转换材料的理想选择。上世纪九十年代,mitzi的研究组首次对有机-无机杂化钙钛矿材料的光电性能进行了探究,发现其具有良好的电子迁移能力,在太阳能电池上具有潜在的应用价值。2009年,kojima课题组首次制备出以ch3hn3pbx3(x=cl,br,i)作为光敏材料的染料敏华太阳能电池,其光电转换效率达到了3.8%。将钙钛矿材料在光电领域的研究引入了快车道。目前,被美国国家可再生能源室(nrel)认证的钙钛矿光伏器件效率已经稳步增长到22.1%。
钙钛矿太阳能电池的研究热潮也带动了基于abx3铅卤钙钛矿量子点(后文写做钙钛矿量子点)发光材料的迅速发展。与正在产业化过程中的ii-vi族量子点相比,钙钛矿量子点具有成本低廉、制备工艺简单、材料毒性低等特点。同时,钙钛矿量子点的发光性能与ii-vi族量子点相当甚至更好:发射光谱覆盖整个可见光波长(410-700nm),荧光量子产率高(>90%),窄发射峰(半峰宽20-50nm)。但是由于钙钛矿材料晶体结构的离子性、配体间的质子交换反应、卤素离子较强的离子迁移能力以及较低的晶体形成能,导致钙钛矿量子点的稳定性较差。能够降低钙钛矿量子点光学性能的外界因素主要有h2o、o2、光和热。晶体结构的离子性使得钙钛矿量子点容易被极性溶剂特别是环境中的h2o降解失去光学性能。o2与h2o配合,加速钙钛矿量子点的分解,同时作为光淬灭剂,降低钙钛矿量子点的光学性能。紫外光则使钙钛矿量子点产生受激辐射,激子非辐射复合过程产生的热效应加速了h2o和o2与钙钛矿量子点的反应过程,同时产生激子热淬灭,降低钙钛矿量子点的光学性能。
针对钙钛矿量子点稳定性差的问题,制备聚合物包覆的钙钛矿量子点复合材料是实现高稳定性钙钛矿量子点的有效方法。yuhuawang课题组将预先合成的cspbx3量子点与聚合物ergo混合,制备出了cspbx3/ergo复合薄膜。该薄膜可以将cspbi3量子点在空气和水中的稳定性从5h延长到25h以上。a.paulalivisatos课题组合成了不同形貌的钙钛矿纳米晶,并将它们分别包入聚月桂基甲基丙烯酸酯(plma),聚苯乙烯-乙烯-丁烯-苯乙烯(sebs)以及聚苯乙烯(ps)中,使得钙钛矿量子点的稳定性得到了巨大的提升,并且钙钛矿纳米线的复合薄膜保持了其偏振发光性能。dwights.seferos课题组将钙钛矿量子点包覆进甲基丙烯酸甲酯(mma),该钙钛矿量子点/mma复合薄膜的稳定性可达到30天以上。然而,基于预先合成的钙钛矿量子点的聚合物复合薄膜普遍存在荧光量子产率不高的问题。cspbx3/ergo复合薄膜的荧光产率只有43%,钙钛矿量子点/mma复合薄膜的荧光量子产率更是从钙钛矿量子点溶液的100%降到了复合材料的56%。这是由于钙钛矿量子点的合成需要繁琐的提纯步骤,如除去反应过程中的大量有机溶剂及长链配体,这个过程会对钙钛矿纳米材料的发光性能造成不利的影响。此外,经过分离提纯的钙钛矿量子点也会影响其在包覆基质中的分散,团聚会使量子点的荧光量子产率急剧下降,并且使得到的复合材料光透过率低,影响器件的性能。为了提高钙钛矿量子点的稳定性同时兼顾其优异的发光性能,研究者开发了原位制备钙钛矿量子点/聚合物复合发光薄膜的方法。yajiedong课题组使用溶胀-去溶胀的方法原位制备了五种不同聚合物基质的钙钛矿量子点/聚合物复合薄膜,其中mapbbr3/ps复合薄膜的荧光量子产率最高达到48%,且能够在水中放置60天而不发生分解。jiuyangzhang课题组在沉淀法制备cspbbr3量子点的过程中加入聚合物,原位制备了cspbbr3/聚甲基丙烯酸甲酯,cspbbr3/聚甲基丙烯酸丁酯和cspbbr3/聚苯乙烯复合材料,cspbbr3/聚甲基丙烯酸甲酯的荧光量子效率为62.4%。但是目前钙钛矿量子点/聚合物复合发光薄膜的原位制备研究主要集中在绿色发光薄膜,红光发射的钙钛矿量子点复合发光薄膜的荧光量子产率和稳定性都比较低,不能满足应用要求。
目前基于钙钛矿量子点的红光发光材料主要是ch3nh3pbi3(mapbi3),nh2chnh2pbi3(fapbi3)和cspbi3,它们块体材料的带隙小于1.73ev。将这些材料制备成量子点后由于量子限域效应其带隙会增大,但是目前得到的全碘钙钛矿量子点的发光波长仍然普遍大于650nm,限制了其在显示等领域的应用。若要进一步增大其带隙,将发射光波长蓝移至620~630nm之间,需要使用br-等进行掺杂。掺杂离子的引入会降低材料的结构稳定性,在长时间使用后混合卤素的钙钛矿量子点会发生相分离,使量子点的发光峰位发生变化,恶化显示性能。因此,需要寻找新的方法调节全碘钙钛矿量子点的带隙。
技术实现要素:
本申请的目的是提出在原位制备γ-cspbi3/聚合物复合发光材料的过程中对所生成的γ-cspbi3量子点表面进行包覆。经过包覆后量子点能带结构以及介电常数发生变化,使其带隙增大,发光峰位蓝移。经过优化制备参数,可以得到发光峰位于610-680nm之间,半峰宽小于32nm,荧光量子产率大于80%并且稳定性高的复合发光薄膜,解决了以往红光全碘cspbi3钙钛矿量子点难于在背光显示等光电子器件中应用的问题。
根据本申请的一个方面,提供了一种复合发光材料,经过包覆/钝化后量子点能带结构以及介电常数发生变化,使其带隙增大,发光峰位蓝移。
所述复合发光材料,其特征在于,所述复合发光材料包括钙钛矿纳米材料和基质;
所述钙钛矿纳米材料包括γ-cspbi3和添加元素m;
所述添加元素m选自li、na、k、rb中的至少一种。
可选地,所述复合发光材料为复合发光薄膜、复合发光块体或复合发光板材。所述复合发光材料可以加工成型成任意需要的形状。
可选地,所述钙钛矿纳米材料分散在所述基质中。
可选地,所述钙钛矿纳米材料为核壳结构;
内核为γ-cspbi3;
表面包括添加元素m。
可选地,所述钙钛矿纳米材料为核壳结构;
内核为γ-cspbi3;
表面包括mpbx3、m4pbx6中的至少一种,其中m选自li、na、k、rb中的至少一种,x选自卤族元素中的至少一种。
可选地,所述钙钛矿纳米材料为核壳结构;
内核为γ-cspbi3;
表面为rbpbi3。
可选地,所述复合发光材料为红色发光材料。
可选地,所述复合发光材料的发光峰在600~680nm。
可选地,所述复合发光材料的发光峰在620~635nm。
可选地,所述钙钛矿纳米材料中γ-cspbi3和添加元素m的摩尔比为1:0.01~10。
可选地,所述钙钛矿纳米材料中γ-cspbi3和添加元素m的摩尔比为1:0.2~0.5。
可选地,所述钙钛矿纳米材料中的γ-cspbi3的形貌选自γ-cspbi3量子点、γ-cspbi3纳米片、γ-cspbi3纳米线中的至少一种。
可选地,所述钙钛矿纳米材料在至少一个维度上的尺寸为2~50nm。
可选地,所述钙钛矿纳米材料中的γ-cspbi3为γ-cspbi3量子点颗粒;
所述γ-cspbi3量子点颗粒的平均粒径为14nm。
可选地,所述基质为聚合物。
可选地,所述聚合物选自聚偏氟乙烯、聚偏氟乙烯和三氟乙烯共聚物、聚丙烯腈、聚醋酸乙烯酯、醋酸纤维素、氰基纤维素、聚砜、芳香聚酰胺、聚酰亚胺、聚碳酸酯、聚苯乙烯、聚甲基丙烯酸甲酯中的至少一种。
可选地,所述钙钛矿纳米材料与所述基质的质量比为1:1~100。
可选地,所述基质与钙钛矿纳米材料的质量比为1:1~30。
可选地,所述复合发光材料还包括添加剂,所述添加剂分散在所述基质中;
所述添加剂选自溴化锌、碘化锌、溴化亚锡、碘化亚锡、溴化镉、碘化镉中的至少一种。
可选地,所述基质与添加剂的质量比为1:0.001~0.5。
可选地,所述钙钛矿纳米材料还包括表面配体,所述表面配体形成在γ-cspbi3的表面;
所述表面配体含有有机酸、有机酸卤代物、c4~c24有机胺、c4~c24有机胺的卤代物中的至少一种。
可选地,pbi2+csi+m的质量之和与所述表面配体的质量比为1:0.001~1。
可选地,所述复合发光材料为复合发光薄膜;
所述复合发光薄膜的厚度为0.001-5mm。
具体地,γ-cspbi3钙钛矿纳米材料为γ-cspbi3钙钛矿量子点;所述γ-cspbi3量子点在至少一个维度上的尺寸不大于20nm。
所述γ-cspbi3量子点具有内核,所述内核的结构式为cspbi3,其中pb和i构成配位八面体结构,cs填充在所述八面体结构的间隙中形成的正交结构相。γ-cspbi3的晶体结构中,其键角α=β=γ=90°,键长
根据本申请的另一个方面,提供一种上述的复合发光材料的制备方法。
所述复合发光材料的制备方法,其特征在于,包括以下步骤:
(1)获得含有基质、钙钛矿前驱体、含有添加元素m的化合物的前驱体溶液;
(2)将所述前驱体溶液成型,得到所述改性钙钛矿纳米材料。
可选地,步骤(1)中所述基质为聚合物;
所述聚合物选自聚偏氟乙烯、聚偏氟乙烯和三氟乙烯共聚物、聚丙烯腈、聚醋酸乙烯酯、醋酸纤维素、氰基纤维素、聚砜、芳香聚酰胺、聚酰亚胺、聚碳酸酯、聚苯乙烯、聚甲基丙烯酸甲酯中的至少一种。
可选地,步骤(1)中所述前驱体溶液中还含有溶剂;
所述溶剂选自n,n-二甲基甲酰胺、二甲基亚砜、三甲基磷酸酯、磷酸三乙酯、n-甲基吡咯烷酮、二甲基乙酰胺中的至少一种。
可选地,所述添加元素m来自含有添加元素m的化合物;
所述含有添加元素m的化合物选自licl、nacl、kcl、rbcl、libr、nabr、kbr、rbbr、lii、nai、ki、rbi、li2co3、na2co3、k2co3、rb2co3、li金属有机物、na金属有机物、k金属有机物、rb金属有机物中的至少一种。
可选地,步骤(1)包括:
(s11)获得含有基质的甲溶液;
(s12)获得含有csi、pbi2、添加元素m的乙溶液。
(s13)将甲溶液和乙溶液混合得到所述前驱体溶液。
可选地,所述乙溶液中,pbi2与(csi+m)的摩尔比为1:0.1~3;
csi和m的摩尔比为1:0.01~10;
溶剂:(pbi2+csi+m)的质量比为1:0.001~3。
可选地,所述甲溶液中,基质与溶剂的质量比为1:1~100。
可选地,甲溶液和乙溶液的质量比为1:0.02~5。
可选地,所述甲溶液中,基质与溶剂的质量比为1:1、1:3、1:4、1:5、1:6、1:7、1:8、1:9、1:10、1:12、1:15、1:20、1:30、1:40、1:50、1:60、1:70、1:80、1:90、1:100及任意两个比值之间的范围值。
可选地,乙溶液中,pbi2和(csi+m)的摩尔比为1:0.1、1:0.4、1:0.5、1:0.6、1:0.75、1:0.9、1:1、1:1.1、1:1.5、1:2、1:3及任意两个比值之间的范围值。
可选地,乙溶剂和(pbi2+csi+m)的质量比为1:0.001、1:0.01、1:0.03、1:0.045、1:0.05、1:0.1、1:0.2、1:0.8、1:0.9、1:1、1:3及任意两个比值之间的范围值。
可选地,步骤(s13)中,甲溶液和乙溶液的质量比为1:0.02、1:0.1、1:0.5、1:0.6、1:0.8、1:1、1:2、1:3、1:5及任意两个比值之间的范围值。
可选地,所述甲溶液中还包括添加剂,所述添加剂选自溴化锌、碘化锌、溴化亚锡、碘化亚锡、溴化镉、碘化镉中的至少一种。
可选地,所述基质与添加剂的质量比为1:0.001~0.5。
可选地,所述甲溶液中,基质与添加剂的质量比为1:0.001、1:0.003、1:0.01、1:0.015、1:0.4、1:0.5及任意两个比值之间的范围值。
可选地,所述前驱体溶液中还含有表面配体;
所述表面配体含有有机酸、有机酸卤代物、c4~c24有机胺、c4~c24有机胺的卤代物中的至少一种;
所述表面配体在步骤(s12)中添加。
可选地,所述前驱体溶液中,(pbi2+csi+m)与表面配体的质量比为1:0.001~5。
可选地,所述乙溶液中,(pbi2+csi+m)与表面配体的质量比为1:0.001、1:0.015、1;0.01、1:0.02、1:0.1、1:0.6、1:1、1:2、1:3、1:4、1:5及任意两个比值之间的范围值。
可选地,步骤(2)中所述成型包括:
将所述前驱体溶液转移至模板上,成型,得到所述复合发光材料。
可选地,所述转移包括旋涂法、浸渍提拉法、静电纺丝法、溶液下沉法、喷涂法、刮膜法、浇铸法中的至少一种。
可选地,
步骤(2)中所述成型包括干燥;
所述干燥的条件包括:温度80~180℃,时间0.1~48h。
可选地,所述干燥的条件还包括:压力0.01~0.1mpa。
可选地,所述干燥的压力的上限选自0.02mpa、0.03mpa、0.04mpa、0.05mpa、0.06mpa、0.07mpa、0.08mpa、0.09mpa或0.1mpa;下限选自0.01mpa、0.02mpa、0.03mpa、0.04mpa、0.05mpa、0.06mpa、0.07mpa、0.08mpa或0.09mpa。
可选地,所述干燥的温度的上限选自90℃、100℃、110℃、120℃、130℃、140℃、150℃、160℃、170℃或180℃;下限选自80℃、90℃、100℃、110℃、120℃、130℃、140℃、150℃、160℃或170℃。
可选地,所述干燥的时间的上限选自1h、2h、3h、4h、5h、6h、8h、10h、15h、24h、28h、32h、35h、40h或48h;下限选自0.1、0.5h、1h、2h、3h、4h、5h、6h、8h、10h、15h、24h、28h、32h、35h或40h。
具体地,所述改性钙钛矿纳米材料的制备方法,其特征在于,包括以下步骤:
(1)将基质聚合物材料溶解于溶剂中:
有机溶剂包括n,n-二甲基甲酰胺(dmf)、二甲基亚砜(dmso)、三甲基磷酸酯(tmp)、磷酸三乙酯(tep)、n-甲基吡咯烷酮(nmp)、二甲基乙酰胺(dmac)中的至少之一。基质是由有机聚合物构成的,聚合物可以为聚偏氟乙烯(pvdf)、聚偏氟乙烯和三氟乙烯共聚物(p(vdf-trfe))、聚丙烯腈(pan)、聚醋酸乙烯酯(pvac)、醋酸纤维素(ca)、氰基纤维素(cna)、聚砜(psf)、芳香聚酰胺(pa)、聚酰亚胺(pi)、聚碳酸酯(pc)、聚苯乙烯(ps)、聚甲基丙烯酸甲酯(pmma)的至少之一。基质与有机溶剂的质量比为1:(1-50)。将基质材料与有机溶剂混合,机械搅拌直至基质材料完全溶解于有机溶剂中生成澄清透明的溶液。
聚合物基质在γ-cspbi3量子点/聚合物复合发光材料中主要起三个方面的作用:第一,聚合物基质在γ-cspbi3量子点原位生成的过程中对γ-cspbi3量子点的尺寸起到限制的作用。由于聚合物基质的存在,生成的γ-cspbi3量子点之间被隔离开,不能继续生长成为大的颗粒。最终将γ-cspbi3量子点的颗粒尺寸限制在20nm以下。第二,聚合物基质对γ-cspbi3量子点的相变起到限制作用。当γ-cspbi3量子点/聚合物复合薄膜由制备温度降至室温时,cspbi3量子点有由γ相自发转变为δ相的趋势。但是在γ相转变为δ相的过程中,cspbi3的晶胞体积会增大。此时,聚合物给予γ-cspbi3量子点的空间并没有增加,限制了γ-cspbi3量子点转变为δ-cspbi3,使得室温时聚合物复合薄膜中的cspbi3仍为γ相。第三,聚合物基质能够隔绝cspbi3量子点与h2o、o2的接触,使cspbi3量子点不容易分解进而失去光学活性,增强了γ-cspbi3量子点/聚合物复合薄膜的稳定性。
甲溶液的制备过程采用高速搅拌器进行分散。由此,可以进一步提高甲溶液的均匀性和分散性,进而可以提高复合材料的效果。甲溶液可以是通过下列步骤制备的:将基质和添加剂溶解于有机溶剂中,基质与有机溶剂的质量比为1:(1-30),基质与添加剂的质量比为1:(0.01-0.5),机械搅拌混合12h,使基质和添加剂完全溶解在有机溶剂中,得到澄清透明的溶液,获得甲溶液。
(2)获得乙溶液
在该步骤中,将pbi2、csi和添加元素m溶解在有机溶剂中,获得乙溶液。乙有机溶剂包括选自dmf、dmso、tmp、tep、nmp、dmac中的个少之一,且乙有机溶剂与甲有机溶剂混溶。需要说明的是,术语“混溶”特指当将甲有机溶剂与乙有机溶剂混合时,混合溶液不出现分层现象。由此,可以将使甲溶液以及乙溶液混合形成统一的有机溶剂体系,也即是说,甲溶液以及乙溶液中溶解的pbi2、csi、添加元素m、表面配体、聚合物基质、添加剂等原料组分在甲有机溶剂以及乙有机溶剂中的溶解度无显著差异,在宏观和微观结构上均没有出现相分离。其中,pbi2与(csi+m)的摩尔比可以为1:(0.1~3),乙有机溶剂与(pbi2+csi+m)的质量比可以为1:(0.001~1)。
为了进一步提高利用该方法获得的复合发光材料的性能,溶液中加入了有机配体。表面配体为有机酸、长链有机胺或者它们的卤化物。具体的,有机酸可以包括碳原子数为至少为3的饱和烷基酸或不饱和烷基酸;长链有机胺可以为4~24个碳原子的烷基胺胺或芳香胺;所述有机酸或者有机胺的卤化物为所述有机酸或者有机胺对应的卤化物。前驱体溶液中,pbi2+csi+m与有机配体的质量比为1:(0.001~1)。
有机配体的加入能够消除生成的γ-cspbi3量子点表面的缺陷,使非辐射复合减少,增强γ-cspbi3量子点的荧光量子产率。此外,有机配体与γ-cspbi3量子点不同晶面的结合能不同,由此可以控制γ-cspbi3的生长方向,进而达到调控所生成γ-cspbi3形貌(量子点、纳米片、纳米线)的目的。
为了进一步提高利用该方法获得的复合发光材料的性能,溶液中还可以加入添加剂。添加剂包括溴化锌、碘化锌、溴化亚锡、碘化亚锡、溴化镉、碘化镉的至少之一,基质与添加剂的质量比可以为1:(0.001-0.5)。
加入添加剂,能够降低γ-cspbi3量子点的成核速率,使生成的γ-cspbi3量子点尺寸更加均匀,进而得到半峰宽更窄的γ-cspbi3量子点/聚合物复合薄膜。其次,添加剂在γ-cspbi3的生成过程中起到助熔剂的作用,使γ-cspbi3的生成温度由175℃降低至80℃,减少了聚合物薄膜的加工难度。(这一部分添加剂仍然可以有,加入后发光峰位置不会发生移动,但是半峰宽会进一步变窄、发光强度以及发光效率都会提高。)
为了调控所得γ-cspbi3量子点的带隙,原料中加入了包覆剂。包覆剂包括碱金属卤化物(lix、nax、kx、rbx,x=cl、br、i)、碱金属碳酸盐(li2co3、na2co3、k2co3、rb2co3)以及li金属有机物、na金属有机物、k金属有机物、rb金属有机物。由于包覆剂中阳离子的半径小,其与pbi2反应生成的产物容忍因子小于0.8,不能生成钙钛矿结构的材料,因此包覆剂阳离子不能掺入cspbi3量子点中生成掺杂量子点而只能包覆并钝化cspbi3量子点表面,从而增大cspbi3量子点的量子限域效应和介电限域效应,最终使得cspbi3量子点的带隙增大,发光峰蓝移。此外,cspbi3量子点的表面被无机材料包覆,能够进一步增强cspbi3量子点的稳定性,推进该聚合物复合薄膜的实际应用。
(3)形成前驱体溶液
根据本发明的实施例,在该步骤中,将甲溶液与乙溶液混合,获得前驱体溶液。具体的,甲溶液与乙溶液的质量比为1:(0.02~5),机械搅拌2h,获得前驱体溶液。
(4)转移
在该步骤中,将混合均匀的前驱体溶液通过合适的方法转移到模板上,以便形成不同形状的复合材料。其中,模板可以为具有特定形状的模具或者基底。关于模板的具体情况,本领域技术人员可以根据实际应用中对复合发光材料形状的具体要求进行设计。具体的,前驱体溶液转移到辈底或模具上的方法可以包括旋涂法、浸渍提拉法、静电纺丝法、溶液下沉法、喷涂法、刮膜法或浇铸法。由此,可以简便地获得具有薄膜等形状的复合发光材料。
(5)干燥
在该步骤中,干燥具有前驱体溶液的所述模板,以便获得所述复合发光材料。具体的,可以将附着有前驱体溶液的模板放置在真空干燥箱中,在一定条件下脱去前驱体溶液中的有机溶剂,由此,可以控制该有机溶剂体系的挥发条件来控制所述基质的结晶、添加剂的排布、γ-cspbi3量子点颗粒的形核与生长,从而提高复合材料的性能。例如,根据本发明的具体实施例,真空干燥箱中的气压可以在0.01~0.1mpa之间,温度可以在80~180℃之间,干燥处理0.1~48h,得到基于γ-cspbi3量子点颗粒的复合材料厚度可以为0.001~5mm。在不同的干燥温度下,可以得到不同粒径分布的γ-cspbi3量子点,由此,可以控制所得γ-cspbi3量子点/聚合物复合发光薄膜的发光波长覆盖600~680nm。
根据本申请的又一方面,提供一种半导体器件。
所述半导体器件,其特征在于,含有上述的复合发光材料、根据上述的复合发光材料的制备方法制备的复合发光材料中的至少一种。
可选地,所述半导体器件包括电致发光器件、光致发光器件、太阳能电池、显示器件、传感器件、压电器件、非线性光学器件。
根据本申请的又一方面,提供一种柔性器件。
所述柔性器件,其特征在于,含有上述的复合发光材料、根据上述的复合发光材料的制备方法制备的复合发光材料中的至少一种。
可选地,所述柔性器件包括基底、金属阳极、空穴传输层、发光层、电子传输层和金属阴极;
所述基底含有上述的复合发光材料、根据上述的复合发光材料的制备方法制备的复合发光材料中的至少一种。
根据本申请的又一方面,提供一种双色复合发光材料。
所述双色复合发光材料,其特征在于,包括层叠的绿色发光薄膜和红色发光薄膜;
所述红色发光薄膜含有上述的复合发光材料、根据上述的复合发光材料的制备方法制备的复合发光材料中的至少一种。
可选地,所述红色发光薄膜为rbpbi3包覆γ-cspbi3量子点/聚甲基丙烯酸甲酯复合薄膜。
可选地,所述绿色发光薄膜为ch3nh3pbbr3量子点/聚偏氟乙烯复合薄膜。
根据本申请的又一方面,提供一种lcd显示器。
所述lcd显示器,其特征在于,含有上述的双色复合发光材料中的至少一种。
根据本申请的又一方面,提供一种光致发光器件。
所述光致发光器件,其特征在于,包括蓝光芯片驱动模组、蓝光芯片散热模组和双色复合发光材料;
所述双色复合发光材料选自上述的双色复合发光材料中的至少一种。
本申请能产生的有益效果包括:
1)本申请所提供的包覆/钝化后量子点能带结构以及介电常数发生变化,使其带隙增大,发光峰位蓝移。经过优化制备参数,可以得到发光峰位于600-680nm之间,半峰宽小于32nm,荧光量子产率大于80%并且稳定性高的复合发光薄膜,解决了以往红光全碘cspbi3钙钛矿量子点难于在背光显示等光电子器件中应用的问题;
2)本申请所提供的cspbi3量子点的表面被无机材料包覆,能够进一步增强cspbi3量子点的稳定性,推进该聚合物复合薄膜的实际应用。
附图说明
图1是实施例1制备的γ-cspbi3/rbpbi3核壳结构量子点/聚合物复合发光薄膜的荧光发射光谱。
图2是实施例1制备的γ-cspbi3/rbpbi3核壳结构量子点/聚合物复合发光薄膜的xrd谱图。
图3是实施例2制备的γ-cspbi3/rbpbi3核壳结构量子点/聚合物复合发光薄膜的荧光发射光谱。
图4是实施例2制备的γ-cspbi3/rbpbi3核壳结构量子点/聚合物复合发光薄膜的xrd谱图。
图5是实施例3制备的γ-cspbi3/rbpbi3核壳结构量子点/聚合物复合发光薄膜的荧光发射光谱。
图6是实施例3制备的γ-cspbi3/rbpbi3核壳结构量子点/聚合物复合发光薄膜的xrd谱图。
图7是根据本实施例的柔性电致发光器件结构示意图
图8是根据本申请实施的双色发光复合薄膜结构示意图;
图9根据本申请实施的lcd显示器件背光模组的结构示意图;
图10是根据本申请实施的光致发光器件结构示意图。
具体实施方式
下面结合实施例详述本申请,但本申请并不局限于这些实施例。
如无特别说明,本申请的实施例中的原料均通过商业途径购买。
本申请的实施例中分析方法如下:
利用panalyticalx’pert3粉末衍射仪进行xrd分析。
利用variancary5分光光度计进行透过光谱分析。
利用flsp920荧光光谱仪进行荧光发射光谱分析。
实施例1
(1)将聚合物溶解于有机溶剂中,控制聚合物:有机溶剂质量比=1:3,加入添加剂zncl2,聚合物与zncl2的质量比为1:0.01,机械搅拌不小于6h,使聚合物完全溶解于有机溶剂中得到澄清透明的溶液,此溶液为甲溶液。所述聚合物为聚甲基丙烯酸甲酯(pmma);所述有机溶剂为n,n-二甲基甲酰胺(dmf)。
(2)将pbi2粉末、csi粉末、rbi粉末混合,控制摩尔比为:pbi2:(csi+rbi)=1:1,csi:rbi=1:1。加入有机溶剂,控制质量比为:有机溶剂:(pbi2+csi+rbi)=1:0.045。在加入有机配体溴化辛胺,控制(pbi2+csi+rbi)与有机配体的质量比为1:0.15。混合后机械搅拌6h,得到澄清透明的溶液,此溶液为乙溶液。该步骤中所述有机溶剂为n,n-二甲基甲酰胺(dmf)。
(3)将步骤(1)中所述的甲溶液与步骤(2)中所述的乙溶液混合,控制质量比为:甲溶液:乙溶液=1:0.5,机械搅拌24h,得到均匀混合的前驱体溶液。
(4)将上述步骤(3)中所述的前驱体溶液荣国旋涂法转移到透明玻璃片上,使前驱体溶液分布均匀。通过控制旋涂装置的转速为1500转/分钟旋涂30秒,使前驱体溶液再透明玻璃片上的厚度约为0.05mm。然后把涂覆有前驱体溶液的透明玻璃片置于真空干燥箱中,真空干燥箱的气压为0.1mpa,温度为50℃,放置10min,除去有机溶剂。再将去除溶剂的玻璃片从真空干燥箱中取出,放置在130℃的加热板上30min,γ-cspbi3量子点首先在pmma基质中原位生成,csi消耗完后rbi与pbi2再反应生成rbpbi3并包覆在γ-cspbi3上,得到γ-cspbi3/rbpbi3核壳结构量子点/pmma复合薄膜。标记为样品1。图1为样品1的荧光发射光谱,发光峰位位于621nm,半峰宽32nm,荧光量子产率87%。图2为样品1的xrd图,在13.3°出现的峰为rbpbi3的衍射峰,其他峰为γ-cspbi3的衍射峰。
实施例2
其余步骤同实施例1。所不同的是,在甲溶液中,控制聚合物与有机溶剂的质量比为1:30。在乙溶液中,控制有机溶剂:(pbi2+csi+rbi)的质量比=1:1,csi与rbi的摩尔比为1:0.2。机械搅拌将溶液混合均匀,真空干燥箱去除溶剂后放置在110℃的加热板上30min,获得γ-cspbi3/rbpbi3核壳结构量子点/pmma复合薄膜,记为样品2。图3为样品2的荧光发射光谱,发光峰位位于647nm,半峰宽31nm,荧光量子产率92%。图4样品2的xrd图,在13.3°出现的峰为rbpbi3的衍射峰,其他峰为γ-cspbi3的衍射峰。
实施例3
其余步骤同实施例1。所不同的是,控制聚合物与有机溶剂的质量比为1:10,并加入添加剂cdi2,聚合物与添加剂的质量比为1:0.05。在乙溶液中,控制有机溶剂:(pbi2+csi+rbi)的质量比=1:3,csi与rbi的摩尔比为1:0.01。机械搅拌将溶液混合均匀,真空干燥箱去除溶剂后放置在150℃的加热板上30min,获得γ-cspbi3/rbpbi3核壳结构量子点/pmma复合薄膜,记为样品3。图5为样品3的荧光发射光谱,发光峰位位于678nm,半峰宽34nm,荧光量子产率78%。图6为样品3的xrd图,在13.3°出现的峰为rbpbi3的衍射峰,其他峰为γ-cspbi3的衍射峰。
实施例4
其余步骤同实施例1,所不同的是,在甲溶液中,聚合物为聚丙烯氰(pan),有机溶剂为二甲基亚砜(dmso),聚合物:有机溶剂质量比为1:6,加入添加剂zni2,控制质量比为聚合物:zni2=1:0.5,机械搅拌不小于6h得到澄清透明的溶液。在乙溶液中,有机溶剂为二甲基亚砜(dmso),控制pbi2:(csi+li2co3)的摩尔比为1:0.5,csi与li2co3的摩尔比为1:0.005,控制有机溶剂:(pbi2+csi+li2co3)的质量比为1:0.001。在前驱体溶液中,控制甲溶液:乙溶液的质量比为1:1。将前驱体溶液通过浸渍提拉法转移到透明玻璃片上,控制前驱体溶液再透明玻璃片上的厚度为0.2mm,真空干燥箱中的气压在0.01mpa,温度为30℃,真空干燥1h。再将去除溶剂的玻璃片从真空干燥箱中取出,放置在130℃的加热板上20min,得到γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜。
实施例5
其余步骤同实施例4,所不同的是,控制有机溶剂:(pbi2+csi+li2co3)的质量比为1:0.1,csi与li2co3的摩尔比为1:0.1。机械搅拌将溶液混合均匀,真空干燥箱去除溶剂后放置在150℃的加热板上10min,获得γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜。
实施例6
其余步骤同实施例1,所不同的是,在甲溶液中,聚合物为聚偏氯乙烯(pvdf),有机溶剂为三甲基磷酸酯(tmp),控制聚合物:有机溶剂的质量比为1:15。在乙溶液中,有机溶剂为三甲基磷酸酯(tmp),控制有机溶剂与(pbi2+csi+nabr)的质量比为1:0.2,pbi2与(csi+nabr)的摩尔比为1:0.1,csi与nabr的摩尔比为1:0.001。将甲溶液与乙溶液以质量比1:0.02混合搅拌均匀。将前驱体溶液通过溶液下沉法转移到玻璃培养皿中,控制前驱体溶液再玻璃培养皿中的厚度为5mm,真空干燥箱中气压为0.05mpa,温度为110℃,干燥10h得到γ-cspbi3/napbbr0.1i2.9核壳结构量子点/pvdf复合薄膜。
实施例7
其余步骤同实施例1,所不同的是,在甲溶液中,聚合物聚甲基丙烯酸甲酯(pmma)与n,n-二甲基甲酰胺的质量比为1:10,再加入添加剂cdbr2,控制聚合物基质与添加剂的质量比为1:0.01,机械搅拌混合不小于6h,得到澄清透明的溶液。在乙溶液中,有机溶剂为n,n-二甲基甲酰胺(dmf),控制有机溶剂与(pbi2+csi+rbbr)的质量比为1:0.9,pbi2与csi的摩尔比为1:3,csi与rbbr的摩尔比为1:5。在前驱体溶液中,控制甲溶液:乙溶液的质量比为1:3,机械搅拌18h,得到澄清透明的前驱体溶液。将前驱体溶液下沉转移到玻璃培养皿中,控制前驱体溶液在玻璃培养皿中的厚度为3mm,真空干燥箱中的气压为0.05mpa,温度在150℃,干燥8h,得到γ-cspbi3/rb4pbbr3i3核壳结构量子点/pvdf复合薄膜。
实施例8
其余步骤同实施例7,所不同的是,在甲溶液中,控制聚合物与有机溶剂的质量比为1:4,加入添加剂cdi2,控制聚合物与添加剂cdi2的质量比为1:0.01。机械搅拌不小于6h,得到澄清透明的溶液。在乙溶液中,控制pbi2与(csi+rbcl)的摩尔比为1:0.5,csi与rbcl的摩尔比为1:10,溶剂与(pbi2+csi+rbcl)的质量比为1:0.01,机械搅拌不小于6h,得到澄清透明的溶液。在前驱体溶液中,控制甲溶液与乙溶液的质量比为1:0.1,机械搅拌12h。将前驱体溶液通过静电纺丝法转移到透明的pet片上,控制前驱体溶液在透明pet片上的厚度为2mm,真空干燥箱的气压在0.07mpa,温度在40℃,干燥15min去除有机溶剂。再将去除溶剂的玻璃片从真空干燥箱中取出,放置在80℃的加热板上1h,cspbi3量子点在pmma基质中原位生成,得到γ-cspbi3/rb4pbcl6核壳结构量子点/pmma复合薄膜。
实施例9
其余步骤同实施例8,所不同的是在甲溶液中聚合物为聚砜(psf),添加剂为znbr2,聚合物与添加剂的质量比为1:0.003。在乙溶液中,有机溶剂与(pbi2+csi+rbcl)的质量比为1:0.03。转移前驱体溶液时,控制前驱体溶液在透明pet片上的厚度为0.5mm。获得附在透明pet片上的γ-cspbi3/rbpbi3核壳结构量子点/psf复合薄膜。
实施例10
其余步骤同实施例1,所不同的是,在甲溶剂中,聚合物为聚偏氯乙烯(pvdf),有机溶剂为n,n-二甲基甲酰胺(dmf)。控制聚合物:有机溶剂的质量比为1:15。乙溶剂中有机溶剂为n,n-二甲基甲酰胺(dmf),有机溶剂与(pbi2+csi+n-苯基甘氨酸钾盐)的质量比为1:0.1,pbi2:(csi+n-苯基甘氨酸钾盐)的摩尔比为1:0.6,csi与n-苯基甘氨酸钾盐的摩尔比为1:2。在前驱体溶液中,控制甲溶液与乙溶液的质量比为1:0.1。将前驱体溶液通过喷涂法转移到透明聚碳酸酯(pc)片上,控制前驱体溶液在透明聚碳酸酯(pc)片上的厚度为1mm,真空干燥箱的气压为0.02mpa,温度20℃,干燥30min去除溶剂。再将去除溶剂的玻璃片从真空干燥箱中取出,放置在100℃的加热板上1h,得到附着在透明聚碳酸酯(pc)片上的γ-cspbi3/kpbi3核壳结构量子点/pvdf复合薄膜。
实施例11
其余步骤同实施例1,所不同的是,在甲溶液中聚合物与有机溶剂的质量比为1:7,所述聚合物为质量比为1:1的聚甲基丙烯酸甲酯(pmma)与聚丙烯腈(pan)的混合物。再加入添加剂sni2,基质与添加剂的质量比为1:0.4,机械搅拌不小于6h,得到澄清透明的溶液。乙溶液中,溶剂与(pbi2+csi)的质量比为1:0.8,再加入碘化辛胺作为配体,(pbi2+csi+rbi)与碘化辛胺的质量比为1:0.1,机械搅拌不小于6h,得到澄清透明的溶液。控制甲溶液与乙溶液的质量比为1:2,机械搅拌18h,得到混合均匀的前驱体溶液。将前驱体溶液通过喷涂法转移到透明聚碳酸酯(pc)片上,实现均匀分布,控制前驱体溶液在透明聚碳酸酯(pc)片上的厚度为0.004mm,然后把涂覆有前驱体溶液的透明聚碳酸酯(pc)片置于真空干燥箱中,真空干燥箱中的气压在0.1mpa,温度为50℃,干燥20min去除有机溶剂,得到附着在透明聚碳酸酯(pc)片上的γ-cspbi3/rbpbi3核壳结构量子点/pmma/pan复合薄膜。
实施例12
其余步骤同实施例1,所不同的是在甲溶液中使用的添加剂是snbr2,聚合物基质与snbr2的质量比为1:0.01。在乙溶液中,控制pbi2:csi的摩尔比为1:0.4,表面配体为戊酸,(pbi2+csi+rbi)与戊酸的质量比为1:0.001。在前驱体溶液中,控制甲溶液与乙溶液的质量比为1:2,机械搅拌不小于24h。将前驱体溶液通过浇筑法转移到透明硅胶片上,控制前驱体溶液在透明硅胶片上的厚度为1mm,真空干燥箱中的气压为0.03mpa,温度为100℃,干燥48h,得到附着在透明硅胶片上的γ-cspbi3/rbpbi3核壳结构量子点/pmma复合薄膜。
实施例13
其余步骤同实施例12,所不同的是在甲溶液中聚合物为聚偏氯乙烯(pvdf),有机溶剂与聚偏氯乙烯(pvdf)的质量比为1:7,添加剂为zni2,基质与zni2的质量比为1:0.015。乙溶液中pbi2与(csi+rbbr)的摩尔比为1:1.1,csi与rbbr的摩尔比为1:10,,添加的表面配体为3,5-二甲基苯胺,(pbi2+csi+rbbr)与3,5-二甲基苯胺的质量比为1:0.1。控制甲溶液与乙溶液的质量比为1:1。得到附着在透明硅胶片上的γ-cspbi3/rb4pbbr6核壳结构量子点/pvdf复合薄膜。
实施例14
其余步骤同实施例1,所不同的是在甲溶液中加入添加剂sni2,基质pmma与sni2的质量比为1:0.4。乙溶液中pbi2与(csi+licl)的摩尔量比为1:0.9,csi与licl的摩尔比为1:0.005,再加入表面配体碘化十二胺,(pbi2+csi+licl)与碘化十二胺的质量比为1:1。涂覆后所放置热台的温度为120℃,加热时间40min,得到γ-cspbi3/lipbcl3核壳结构量子点/pmma复合薄膜。
实施例15
其余步骤同实施例1,所不同的是甲溶液中聚合物基质与有机溶剂的质量比为1:10,聚合物为聚碳酸酯(pc),有机溶剂为n,n-二甲基甲酰胺(dmf)。乙溶剂中添加的表面配体为乙酸和十二胺,乙酸与十二胺的质量比为1:3,(pbi2+csi+rbi)与表面配体的质量比为1:0.02,csi与rbi的摩尔量比为1:8。前驱体溶液中甲溶液与乙溶液的质量比为1:0.8。控制加热板的加热温度为120℃,烘干30min,得到γ-cspbi3-rbi异质结结构量子点/pc复合薄膜。
实施例16
其余步骤同实施例1,所不同的是甲溶液中的聚合物基质为聚苯乙烯(ps),基质与有机溶剂的质量比为1:20,有机溶剂为n,n-二甲基甲酰胺(dmf)。乙溶液中加入的表面配体为溴化辛胺,(pbi2+csi+kbr)与表面配体溴化辛胺的质量比为1:0.6,csi与kbr的摩尔量比为1:10。前驱体溶液中甲溶液与乙溶液的质量比为1:0.6。将前驱体溶液通过旋涂法转移到透明石英玻璃片上,控制前驱体溶液在透明石英玻璃片上的厚度为1mm,真空干燥箱中的气压为0.1mpa,温度为130℃,干燥72h,得到γ-cspbi3-k4pbbr6异质结结构量子点/ps复合材料。
实施例17
其余步骤同实施例1,所不同的是甲溶液中控制聚合物与有机溶剂的质量比为1:10,聚合物为聚偏氯乙烯(pvdf),有机溶剂为二甲基乙酰胺(dmac)。在乙溶液中,控制pbi2:csi的摩尔量比为1:2,有机溶剂为二甲基乙酰胺(dmac),有机溶剂与(pbi2+csi+rbi)的质量比为1:1.5,有机配体为戊酸和3-乙烯基乙胺,戊酸与3-乙烯基乙胺的质量比为1:5,(pbi2+csi)与有机配体的质量比为1:0.01。将前驱体溶液通过旋涂法转移到ito玻璃上,控制前驱体溶液在ito玻璃上的厚度为0.1mm,真空干燥箱中的气压在0.02mpa,温度为40℃,干燥15min,去除有机溶剂。将去除有机溶剂的ito玻璃片放置在130℃的加热板上烘烤45min,得到棒状γ-cspbi3/rbpbi3核壳结构量子点/pvdf复合材料。
实施例18
本申请中的半导体器件可以为柔性器件,结构示意图如图7所示,上述复合发光材料可以为薄膜,并直接用于形成电致发光器件中的柔性透明基底;该柔性器件中还可以进一步具有电致发光材料构成的发光层,将γ-cspbi3量子点颗粒的光致发光性质和电致发光的发光结合在一起,从而可以进一步提高该柔性器件的发光性能。本领域技术人员能够理解的是,上述柔性器件中还可以进一步包括用以实现其器件性能的结构,如图9中所示出的金属阴极、金属阳极、电子传输层、空穴传输层等,在此不再赘述。
实施例19
以实施例5所制备的γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜为基础,制备高色域的白光led发光材料,具体步骤为:
(1)ch3nh3pbbr3量子点/pvdf绿光发射复合薄膜材料的制备
第一溶液中聚合物:有机溶剂的质量比=1:5,聚合物为聚偏氟乙烯(pvdf),有机溶剂为n,n-二甲基甲酰胺(dmf)。机械搅拌12h,得到澄清透明的溶液。第二溶液中pbbr2与ch3nh3br的摩尔比为1:1,有机溶剂:pbbr2的质量比为1:0.01,有机溶剂为n,n-二甲基甲酰胺(dmf)。机械搅拌12h,得到澄清透明的溶液。控制第一溶液与第二溶液的质量比为1:0.2,机械搅拌24h,得到混合均匀的前驱体溶液。
(2)将上述步骤(1)中所述的前驱体溶液通过旋涂的方法转移到透明的pet薄膜上,控制前驱体溶液在透明pet薄膜上的厚度为0.5mm,然后把附着有前驱体溶液的pet透明薄膜置于真空干燥箱中,真空干燥箱气压为0.1mpa,温度为30℃,干燥48h,得到ch3nh3pbbr3量子点/pvdf绿光发射复合薄膜。
(3)将制备好的ch3nh3pbbr3量子点/pvdf绿光发射复合薄膜与实施例5中所制备的γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜复合薄膜进行组合,应用到白光led器件结构中,得到高色域的白光led器件,图8为所述双色发光薄膜的结构示意图。
本实施例中使用的ch3nh3pbbr3量子点/pvdf绿光发射复合薄膜材料,根据公开号为wo2016180364a1,发明名称为“钙钛矿/聚合物复合发光材料、制备方法及用途”的发明专利公开的方法合成得到,也可以由北京理工大学提供。
实施例20
以实施例5所制备的γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜为基础,制备高色域的白光led发光材料,具体步骤为:
在制备好的γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜一侧涂覆有机胶水,与实施例19中步骤(2)中制备好的ch3nh3pbbr3量子点/pvdf复合薄膜ch3nh3pbbr3量子点薄膜一侧贴合,在50℃烘干1h,使胶水固化,得到红光和绿光双光色发射的复合材料。
实施例21
其余步骤同实施例19,所不同的是ch3nh3pbbr3量子点/pvdf复合薄膜的pet基体一侧与胶水粘贴,烘干后得到红光和绿光双光色发射的复合材料。
实施例22
以实施例5所制备的γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜复合材料为基础,制备高色域的白光led发光材料,具体步骤为:
将实施例19步骤(1)中制备的前驱体溶液通过旋涂的方法涂覆在制备好的γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜量子点膜一侧,然后把附着有ch3nh3pbbr3量子点/pvdf复合薄膜前驱体溶液的cspbi3量子点/pmma复合材料薄膜置于真空干燥箱中,真空干燥箱气压为0.1mpa,温度为30℃,干燥48h,得到红光和绿光双光色发射的复合材料。
实施例23
其余步骤同实施例22,所不同的是在γ-cspbi3/lipbi3核壳结构量子点/pan复合量子点膜一侧涂覆聚碳酸酯(pc)有机溶液,所述溶液的有机溶剂为n,n-二甲基甲酰胺(dmf),有机溶剂与聚碳酸酯(pc)的质量比为1:0.8。将涂覆聚碳酸酯(pc)有机溶液的γ-cspbi3/lipbi3核壳结构量子点/pan复合量子点膜置于真空干燥箱中,真空干燥箱气压为0.1mpa,温度为30℃,干燥48h,得到具有聚碳酸酯(pc)薄膜阻隔的cspbi3量子点/pmma复合材料量子点膜。再将实施例19步骤(1)中制备的前驱体溶液通过旋涂的方法涂覆在制备好的γ-cspbi3/lipbi3核壳结构量子点/pan复合量子点膜聚碳酸酯(pc)阻隔膜一侧,然后把附着有ch3nh3pbbr3量子点/pvdf复合薄膜前驱体溶液的cspbi3量子点/pmma复合材料薄膜置于真空干燥箱中,真空干燥箱气压为0.1mpa,温度为30℃,干燥48h,得到红光和绿光双光色发射的复合材料。
实施例24
上述复合发光材料还可以应用于lcd显示器件。具体的,参考图9和10。首先,将γ-cspbi3量子点/聚合物复合红光薄膜与钙钛矿量子点/聚合物复合绿光薄膜结合,制备出双色(红光和绿光)发光薄膜。再将该双色发光薄膜插入到lcd背光模组的多层膜结构之间,也可以直接将上述复合发光材料涂膜到lcd背光模组中的导光板、扩散膜或棱镜膜的上表面或下表面上,从而可以实现蓝光led作为光源的高色域lcd背光模组。
实施例25
以实施例5所制备的γ-cspbi3/lipbi3核壳结构量子点/pan复合量子点膜为基础,制备液晶显示器(lcd)用高色域的背光源,以42英寸的lcd为例,具体步骤为:
(1)42英寸cspbi3量子点/pmma复合发光薄膜的制备
按照实施例5的实验方案配制所需质量的前驱体溶液,用刮膜机将所述的前驱体溶液均匀转移到对应尺寸的玻璃基板上,控制前驱体溶液的厚度为0.2mm,然后将韩有前驱体溶液的玻璃板置于真空干燥箱中,在0.05mpa、150℃下烘干6h,取出待用。然后利用膜转移技术将制备的cspbi3量子点/pmma复合发光薄膜转移到lcd背光模组中的导光板、扩散膜或者棱镜膜上,为了缩减工艺,也可以直接将上述前驱体溶液通过刮膜机转移到lcd背光模组的导光板、扩散膜或者棱镜膜上,再通过相同的条件进行干燥,形成一体的发光层。
(2)42英寸cspbi3量子点/pmma发光层的制备
按照实施例5的实验方案配制前驱体溶液,用刮膜机将所述的前驱体溶液均匀转移到基底上,此处使用的基底包括玻璃板或lcd背光模组的导光板、扩散膜、棱镜膜上,控制前驱体溶液的厚度为0.1mm,置于真空干燥箱中,在0.05mpa、150℃下烘干6h,取出得到高发光效率的cspbi3量子点/pmma红光发射复合薄膜。
(3)lcd背光模组的组装
将步骤(1)和(2)中得到的发光膜插入到lcd背光模组中,可以将lcd背光模组的光源替换为蓝光光源。蓝光光源经过导光板后再通过红色发光层和绿色发光层,最后形成红绿蓝三基色复合的白光。
实施例26
本实施例以钙钛矿/聚合物复合发光材料为基础,制备压电器件,具体步骤为:
(1)按照实施例8的实验方案配制前驱体溶液,然后将所述的前驱体溶液均匀涂覆到基底上,此处使用的基底包括ito导电玻璃或者表面镀金/银的pet、pc柔性聚合物基底。控制前驱体溶液的厚度为0.5mm,置于真空干燥箱中,在0.05mpa、150℃下烘干6h,取出得到高发光效率的γ-cspbi3/rb4pbcl6核壳结构量子点/pmma红光发射复合薄膜。
(2)在制备的γ-cspbi3/rb4pbcl6核壳结构量子点/pmma红光发射复合薄膜表面上镀金电极或者银电极,然后在电极上方涂覆一层保护层,得到简易压电器件原型,通过导线将基于复合薄膜的压电器件的两极接到示波器上。
(3)在制备的基于复合薄膜的压电器件上施加周期性的作用力,可以在示波器上看到周期性的脉冲压电信号。
实施例27
以实施例5所制备的γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜为基础,制备太阳能聚光器,以400平方厘米的聚光器为例,具体步骤为:
(1)400平方厘米γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜的制备
按照实施例5的实验方案配制所需质量的前驱体溶液,用刮膜机将所述的前驱体溶液均匀转移到对应尺寸的玻璃基板上,玻璃基板的厚度为2mm,长、宽均为20cm。控制前驱体溶液的厚度为0.2mm,然后将含有前驱体溶液的玻璃板置于真空干燥箱中,在0.05mpa、150℃下烘干6h,取出待用。
(2)聚光器的制备
将步骤(1)中涂覆有γ-cspbi3/lipbi3核壳结构量子点/pan复合薄膜的玻璃板放置于镀膜机中,给玻璃板的三个侧面镀铝,所镀铝膜的厚度为2μm。取出镀完铝的玻璃板,将条状多晶硅太阳能电池板组装到玻璃板未镀铝的侧面。接通太阳能电池板线路,制备出太阳能聚光器。
以上所述,仅是本申请的几个实施例,并非对本申请做任何形式的限制,虽然本申请以较佳实施例揭示如上,然而并非用以限制本申请,任何熟悉本专业的技术人员,在不脱离本申请技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



