
 商标分类
商标分类  商标转让
商标转让 
单细胞冻融裂解的制作方法
 2021-02-02 17:02:14|
2021-02-02 17:02:14| 380|
380| 起点商标网
起点商标网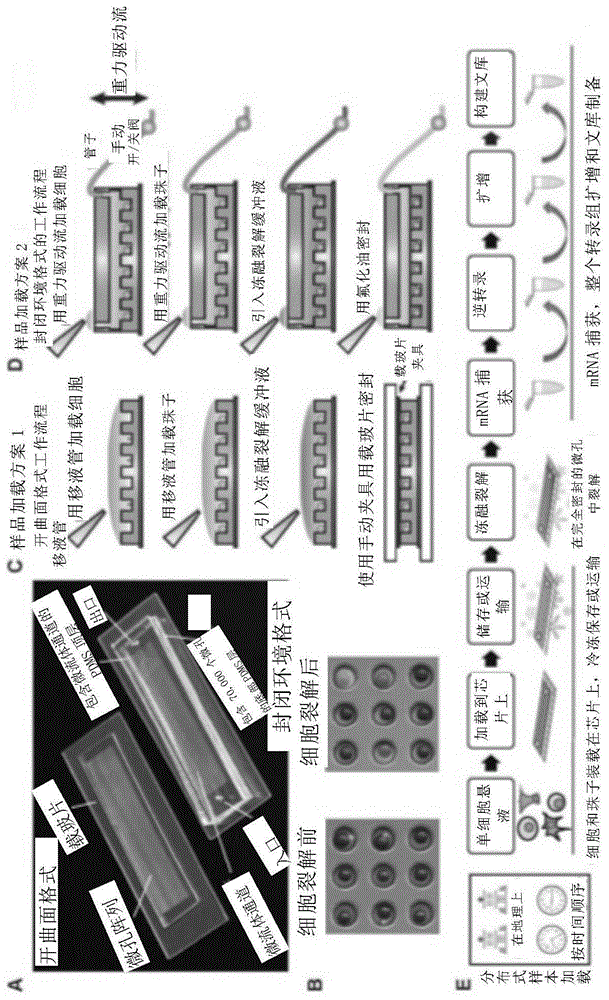
相关申请
基于35u.s.c.§119(e),本申请要求于2018年2月23日提交的美国临时申请号62/634,678的优先权,其通过引用整体并入本文。
联邦赞助的研究
本发明是在美国国立卫生研究院授予ca209992和ca193461以及美国国家科学基金会授予1351443的政府支持下完成的。政府拥有本发明的特定权利。
背景
单细胞rna测序(scrna-seq)成为生物学研究中的主要工具,用于检查复杂样品的异质性、识别不同的细胞亚群,并剖析细胞分化过程和谱系定向。随着分子条形码技术以及将各种微流体平台整合到文库制备步骤中的最新发展,现在可以通过低成本的文库制备对数千个细胞进行测序。
发明概述
在一些实施方式中,本发明提供的是用于直接向3'mrna测序单细胞冻融裂解(即sc-ftdseq)的微芯片平台。该平台通过简化的、可广泛采用的工作流程提供格式灵活性,从而减少了准备步骤的数量和手动操作时间,并提供了高质量的数据。出乎意料的是,本文提供的数据表明,在基于分子条形码的单细胞3'mrna谱分析的背景下,冻融裂解不会裂解该谱分析所需的mrna。不受理论的束缚,这很可能是因为基于分子条形码的单细胞3'mrna谱在3'末端仅需要约50个碱基。令人惊讶的是,单独的冻融裂解步骤(不使用基于去污剂的裂解缓冲液)可有效裂解细胞,并且细胞释放出足够量的mrna,用于单细胞3'mrna转录组分析。
用于单细胞分离和mrna捕获的当前技术(例如微孔阵列)的挑战之一是细胞裂解步骤,其中裂解缓冲液需要以可忽略不计的的物质(例如mrna)损失引入微孔中。另一个挑战是持续不断的工作流程,其要求高水平的实验人员及时完成多个冗长的步骤(至少直到逆转录步骤为止),这对于大多数当前的scrna-seq方法是普遍存在的。将样品的获取和上样与复杂的文库生成步骤分离的功能将有利于研究人员的广泛采用,特别是那些在小型诊所或医疗机构中可能没有scrna-seq库生成经验的人。本文提供了通过实施冻融循环来裂解细胞来极大地改善基于微孔的scrna-seq应用的方法、系统和组合物。出乎意料的是,冻融(一种被认为导致rna片段断裂的不利裂解方法)与单细胞3'mrna测序完全兼容,其在3'末端仅能检测到约50个碱基。与基于去污剂或离液剂的裂解方法相比,例如,本发明的冻融方法不会立即开始裂解,因为在冻融裂解液中没有活性裂解剂,并且消除了对自动快速流体交换或使用半透膜的需求。此外,本发明的方法使细胞上样独立于以下步骤,在一些实施方式中,可以在分布式站点(包括小型诊所或即时医疗点)进行采样(在微孔中捕获细胞/珠),并在集中化设施(运输后)进行下游处理,用于高质量且一致地制备测序文库。这样,scftd-seq可以在一个平台上解决这两个难题,并且可以通过例如用于文库制备和测序的工业化大批量服务,以进一步降低的成本实现scrna-seq的广泛采用。
因此,在某些方面,本文提供了高通量单细胞mrna谱分析方法,该方法包括:将细胞、条形码化的mrna捕获珠和任选的冻融缓冲液上样到微孔阵列上;用盖密封微孔阵列,并产生密封的微孔阵列,其包括上样的细胞和条形码化的mrna捕获珠;冷冻并解冻密封的微孔阵列,从而裂解细胞并从细胞中释放mrna,其中在不存在裂解缓冲液(或任何其他裂解试剂)的情况下裂解细胞;收集条形码化的mrna捕获珠并分离条形码化的珠捕获的mrna;并任选地对mrna进行测序。
在一些方面,本文还提供了高通量单细胞mrna谱分析方法,该方法包括:将细胞、条形码化的mrna捕获珠和任选的冻融缓冲液上样到与微流体通道结合的微孔阵列上,并产生密封的微孔阵列,包括上样的细胞和条码化的mrna捕获珠;冷冻并解冻密封的微孔阵列,从而裂解细胞并从细胞中释放mrna,其中在不存在裂解缓冲液(或任何其他裂解试剂)的情况下裂解细胞;收集条形码化的mrna捕获珠并分离条形码化的珠捕获的mrna;并任选地对mrna进行测序。
本发明还提供了与本文提供的方法一起使用的系统、装置、组合物和试剂盒。
附图的简要说明
图1.scftd-seq平台和工作流程。(a)用于scftd-seq方法的微孔阵列设备。对于封闭环境格式,将微孔阵列结合到微流体通道上。对于开曲面格式,使用微孔阵列,顶部不带通道。(b)在冻融细胞裂解之前(顶部)和之后(底部)在微孔阵列中捕获细胞和珠子。(c)scftd-seq的开曲面格式工作流程。(d)scftd-seq的封闭环境格式工作流程。(e)具有分布式采样的scftd-seq工作流程的示意图。冻融裂解后的文库制备遵循dropseq方法(17)中所述的方案。
图2.scftd-seq的技术性能。(a)混合物种实验揭示了在封闭环境格式下scftd-seq的单细胞分辨率和最小的交叉孔污染(b)开曲面格式也获得了类似的结果。(c)数据下采样后每个细胞检测到的基因数量。以200k读序/细胞的速度,我们分别从人和小鼠细胞中检测到约6,000和约5,500个基因。(d)数据下采样后每个细胞中检测到的转录本数量。以200k读序/细胞的速度,我们分别从人和小鼠细胞中检测到约45,000和约30,00个转录本。(e)人和小鼠细胞的基因数目分布。(f)人和小鼠细胞的转录本数量分布。(g,h)读取scftd-seq的映射质量。(h)对于人类k562细胞,高百分比的读序图映射定位于基因组和外显子区域,而对基因间的、内含子和核糖体rna的映射定位则极少。(h)对于小鼠nih3t3细胞,高百分比的读序图映射定位于基因组和外显子区域,而对基因间的、内含子和核糖体rna的映射定位极少。(i)人k562细胞和小鼠nih3t3细胞的线粒体基因的低百分数表明了高质量的测序文库。误差线代表s.e.m.
图3.鉴定混合样品中的细胞类型。(a)t-sne图,其显示了按1:10比例混合的人k562细胞和小鼠nih3t3细胞的混合物的聚类结果。比对和过滤后鉴定出5,062个细胞,其中451个细胞被推定为人类的,4511个细胞被推定为小鼠的。(b)t-sne图,其显示了以1:1:2比例混合的三种人类细胞系hek-k562-huvec的混合物的测序结果。非监督式聚类分析确定了三个主要聚类。(c)基于细胞类型特异性基因的表达,每个集群可被识别为相应的细胞类型。
图4.推断异质样品(整个肿瘤)中的细胞类型。(a)在yummer1.7同系小鼠模型中生成的与黑色素瘤肿瘤分离的细胞的t-sne图,其中肿瘤细胞表达gfp。鉴定出两个主要集群,其中左边的集群是通过gfp的表达推断的黑素瘤细胞。(b)通过ptprc(cd45)表达推断出免疫细胞集群。(c)非监督式的聚类分析确定了4个集群,将肿瘤和免疫细胞集群分别分为两组。(d)鉴定的集群之间的差异基因表达分析。
图5.循环性的cd4+cxcr5+t细胞的单细胞rna图谱以及sle中的激活状态。(a)t-sne图,其显示了刺激后的与未处理的cd4+cxcr5+pd1低ccr7高t细胞(tcm)的聚类结果。(b)被刺激的和未处理的tcm细胞的差异基因表达分析。(c)在被刺激的和未处理的tcm细胞之间细胞因子基因表达的比较。(d)t-sne图,其显示了刺激后的或未处理的cd4+cxcr5+pd1高ccr7低t细胞(ctfh)的聚类结果。鉴定出与被刺激的和未处理的样品重叠的两个主要集群。(e)被刺激的和未处理的ctfh细胞的差异基因表达分析。(f)在被刺激的和未处理的ctfh细胞之间细胞因子基因表达的比较。(g)被刺激的tcm和ctfh细胞的t-sne图。(h)被刺激的tcm和ctfh细胞的差异基因表达分析。(i)被刺激的tcm和ctfh细胞之间细胞因子基因表达的比较。
发明详述
本发明提供了基于冻融裂解的方法,其产生有效的细胞裂解和mrna捕获,以及高质量的测序文库。该技术在本文中被称为scftd-seq,例如,它使用冻融过程直接裂解含珠微孔中分离的单个细胞,并捕获单细胞来源的mrna,用于转录组测序。scftd-seq与开曲面和封闭环境的细胞上样格式兼容,其因为不需要任何外部设备而具有便携性,并且可以在分布式样本采购现场节省整个文库生成过程。本文中的实施例提供了一个示例,该示例具有在小于10,000个细胞作为输入的情况下每次运行获得约高达5000个单细胞转录组的高性能能力。如本文所示,该方法被应用于包括细胞系和整个肿瘤在内的混合种群的分析,以区分所有主要细胞类型(肿瘤和免疫细胞),还被应用于自身免疫性发病-系统性红斑狼疮患者的发病机制中涉及的循环性滤泡辅助性t细胞和中央记忆t细胞的分析。这些结果提供了对这些稀有t细胞的转录和功能异质性的深入了解,这可能是其在自身免疫中的作用的基础。例如,这些数据证明了scftd-seq是用于大规模并行单细胞3'mrna测序的高性能和称职的技术。它最终可能会将scrna-seq的使用范围从学术实验室扩展到在单细胞测序文库制备方面没有经验的任何潜在用户,并进一步利用工业化的力量来降低成本并确保数据质量,这是未来大规模临床应用的关键一步。
本文提供了单细胞mrna谱分析方法(例如,高通量方法)。在一些实施方式中,该方法包括将细胞(例如,在溶液,例如细胞培养基中)、条形码化的mrna捕获珠和冻融缓冲液上样到微孔阵列上。与用于细胞裂解的缓冲液不同,本发明的冻融缓冲液不包含去污剂或其他裂解试剂,例如tritontmx-100、np-40或十二烷基硫酸钠(sds)。在一些实施方式中,冻融缓冲液包含高渗溶液。在一些实施方式中,冻融缓冲液包括选自以下的至少一种试剂:tris、edta、nacl、dtt和rnase。在一些实施方式中,冻融缓冲液包含tris、edta、nacl、dtt和rnase(例如100mmtris-ph7.5、10mmedta、1mnacl、5μmdtt、0.4u/mlrnase)。在一些实施方式中,冻融缓冲液仅是细胞培养基(不包括任何裂解试剂)。
条形码化的mrna捕获珠可以是聚合物珠。例如,珠可以是琼脂糖珠。珠被认为是条形码的,因为它们与包含独特核苷酸序列的寡核苷酸连接(例如,共价连接)。独特的条形码序列对于特定的mrna捕获珠或mrna捕获珠的亚群可能是唯一的。条码化的mrna捕获珠是本领域已知的(参见,例如,yuanj等人,scirep2016;6:33883,通过引用并入本文)。在一些实施方式中,条形码化的mrna捕获珠的直径为20μm至50μm。例如,条形码化的mrna捕获珠的直径可以为20μm-45μm、20μm-40μm、20μm-35μm、20μm-30μm、20μm-35μm、30μm-50μm、30μm-45μm、30μm-40μm或30μm-35μm。在一些实施方式中,条形码化的mrna捕获珠的直径为20μm、25μm、30μm、35μm、40μm、45μm或50μm。在一些实施方式中,条形码化的mrna捕获珠的直径为35μm。
在一些实施方式中,本发明的微孔阵列包含1,000个微孔至100,000个微孔。例如,微孔阵列可以包含1,000个微孔-10,000个微孔、1,000个微孔–15,000个微孔、10,000个微孔–100,000个微孔或15,000个微孔–100,000个微孔。在一些实施方式中,微孔包括15,000个微孔至70,000个微孔。在一些实施方式中,微孔包含100、1000、2000、3000、4000、5000、6000、7000、8000、9000、10000、15000、20000、25000、30000、35000、40000、45000、50000、55000、60000、65000,70000、75000、80000、85000、90000、95000或10000个微孔。微孔可以是圆形、正方形或矩形。本文涵盖其他微孔形状、腔室和通道。在一些实施方式中,微孔阵列包括直径(宽度)为40μm-60μm或45μm-55μm的微孔。例如,微孔阵列可以包括直径为40μm、50μm或60μm的微孔。
在一些实施方式中,本发明提供的方法是高通量的。因此,可以同时处理许多不同的细胞和/或细胞类型。在一些实施方式中,将100个细胞-20,000个细胞,或1,000个细胞-5,000个细胞上样到微孔阵列上。例如,可以将100、200、300、400、500、600、700、800、900、1000、1500、2000、2500、3000、3500、4000、4500或5000个细胞上样到微孔阵列上。在一些实施方式中,单次运行可以处理5000多个细胞。在一些实施方式中,将10,000或15,000个细胞上样到微孔阵列上。
本发明提供的方法可用于分析多种细胞,包括新鲜分离的原代细胞,例如肿瘤细胞。该方法不受特定细胞类型的限制。
在一些实施方式中,微孔阵列的孔占有率为5%–10%。
在一些实施方式中,阵列微孔的80%以上包含单个条形码的mrna捕获珠。例如,大于85%、大于90%或大于95%的阵列包含单个条形码的mrna捕获珠。在一些实施方式中,80%-100%、80%-95%、80%-90%、80%-85%、85%-100%、85%-95%、85%-90%、90%-100%或90%-95%的阵列微孔包含单个条形码的mrna捕获珠。
在一些实施方式中,本发明的方法还包括用盖密封微孔阵列,并产生包含上样的细胞、条形码化的mrna捕获珠和冻融缓冲液的密封微孔阵列。在一些实施方式中,使用手动夹具用载玻片密封微孔。在一些实施方式中,将微孔阵列密封而不使用半透(纳米多孔)膜。在一些实施方式中,如上所述,将细胞上样到开放的微孔阵列上,然后用盖,如盖玻片密封。然而,在其他实施方式中,将细胞、条形码化的mrna捕获珠和冻融缓冲液上样到与微流体通道结合的封闭微孔阵列上(参见例如实施例部分)。
在一些实施方式中,本发明的冷冻和解冻(冻融)步骤包括冷冻密封的微孔阵列的细胞(例如,在-80℃),然后解冻细胞(例如,在4℃下或在室温(25℃)),以使细胞的细胞壁破裂并从细胞中释放出mrna。在一些实施方式中,该方法包括至少一个、至少两个或至少三个冻融循环。在一些实施方式中,该方法包括三个冻融循环。
在冻融循环之后,可将微阵列孵育一段时间(例如30-90分钟,例如60分钟),使得条形码的mrna捕获珠捕获释放的mrna。mrna的polya尾巴将与条形码捕获珠的polyt(oligo(dt))序列结合。
在一些实施方式中,该方法进一步包括收集条形码化的mrna捕获珠(例如,将微孔阵列倒置并使阵列离心)和分离由条形码化的珠捕获的mrna。
在一些实施方式中,分离的mrna是序列,例如使用本领域已知的任何测序方法所获得的序列。
本发明进一步提供了由以下编号的段落涵盖的实施方式:
1.一种高通量单细胞mrna谱分析方法,该方法包括:
将细胞、条形码化的mrna捕获珠以及任选的冻融缓冲液上样到微孔阵列上;
用盖密封微孔阵列,并产生密封的微孔阵列,其包括上样的细胞和条形码化的mrna捕获珠;
冷冻并解冻密封的微孔阵列,从而裂解细胞并从细胞中释放mrna,其中在不存在裂解缓冲液(或任何其他裂解试剂)的情况下裂解细胞;
收集条形码化的mrna捕获珠并分离条形码化的珠捕获的mrna;和
对mrna进行测序。
2.一种高通量单细胞mrna谱分析方法,该方法包括:
将细胞、条形码化的mrna捕获珠和任选的冻融缓冲液上样到与微流体通道结合的微孔阵列上,并产生包含上样的细胞和条形码化的mrna捕获珠的密封微孔阵列;
冷冻并解冻密封的微孔阵列,从而裂解细胞并从细胞中释放mrna,其中在不存在裂解缓冲液(或任何其他裂解试剂)的情况下裂解细胞;
收集条形码化的mrna捕获珠并分离条形码化的珠捕获的mrna;和
对mrna进行测序。
3.一种方法,包括:
将细胞、条形码化的mrna捕获珠和冻融缓冲液上样到微孔阵列上;
用盖密封微孔阵列,并产生密封的微孔阵列,其包括上样的细胞、条形码化的mrna捕获珠和冻融缓冲液;和
冷冻并解冻密封的微孔阵列,从而从细胞中释放mrna。
4.一种方法,包括:
将细胞、条形码化的mrna捕获珠和冻融缓冲液上样到与微流体通道结合的微孔阵列上,并产生包含上样的细胞、条形码化的mrna捕获珠和冻融缓冲液的密封微孔阵列;和
冷冻并解冻密封的微孔阵列,从而从细胞中释放mrna。
5.如段3或4所述的方法,其进一步包括收集条形码化的mrna捕获珠并分离由条形码化的珠捕获的mrna。
6.如段5所述的方法,其进一步包括对mrna测序。
7.如段落1-6中任一段所述的方法,其中所述条形码化的mrna捕获珠的直径为20μm-50μm或35μm。
8.如段1-7中任一段所述的方法,其中微孔阵列包括直径(宽度)为40μm-60μm或50μm的微孔。
9.如段1-8中任一段所述的方法,其中微孔阵列包括1,000个微孔-100,000个微孔,或15,000个微孔-70,000个微孔。
10.如段1-9中任一段所述的方法,其中将100个细胞-20,000个细胞,或1,000个细胞-5,000个细胞上样到微孔阵列上。
11.如段1-10中任一段所述的方法,其中所述微孔阵列以5%至10%的孔占有率上样。
12.如段1-11中任一段所述的方法,其中大于80%或大于90%的阵列微孔包含单个条形码的mrna捕获珠。
13.如段1-12中任一段所述的方法,其中所述条形码化的mrna捕获珠通过离心收集。
14.如段1-13中任一段所述的方法,其中所述冻融缓冲液不包含去污剂(例如tritonx-100、np-40或sds)或其他裂解试剂。
15.如段1-14中的任一段所述的方法,其中,所述冻融缓冲液包括高渗溶液。
16.如段1-15中任一段所述的方法,其中所述冻融缓冲液包含选自tris、edta、nacl、dtt和rnase的至少一种试剂。
17.如段落16所述的方法,其中所述冻融缓冲液包含tris、edta、nacl、dtt和rnase。
18.如段1-17中任一段所述的方法,其中将微孔阵列密封而不使用半透(纳米多孔)膜。
19.如段落1-18中任一段所述的方法,其中所述冻融步骤包括将密封的微孔阵列在-80℃下冷冻。
20.如段1-19中的任一段所述的方法,其中所述冻融步骤被重复至少两次。
21.如段1-20中任一段所述的方法,其中所述细胞是分离的原代细胞。
22.如段1-21中任一段所述的方法,其中所述条形码化的mrna捕获珠与包含用于mrna捕获的条形码核苷酸序列和oligo(dt)区域的寡核苷酸共价连接。
实施例
实施例1.scftd-seq工作流程
scftd-seq利用微孔阵列作为共分离单细胞的平台,并在冻融细胞裂解之前使用独特条形码的mrna捕获珠(数据未显示)。微孔阵列的设计与先前所述的类似(22,23),并且阵列由聚二甲基硅氧烷(pdms)制成。微孔的大小取决于mrna捕获珠的大小(约35μm);我们使用的直径约为45-55μm,高度约为50μm,以确保仅容纳单个珠子以及大多数哺乳动物细胞类型。这种尺寸的选择还有助于在mrna捕获后通过倒置设备后进行吹扫(用于封闭环境的细胞接种)或离心(用于开曲面的细胞接种)直接去除珠子。微孔阵列的通量由孔总数决定,并且非常具有可扩展性。在这项研究中,我们制造了具有15,000-70,000孔的阵列,从而能够在单次运行中对约1,000-5,000个细胞进行测序,其中阵列的孔占用率为5-10%,以最大程度地减少双重占用(细胞二倍体)。在开曲面上样格式(图1a)中,高通量有所不同,但主要取决于阵列上使用的面积以及要上样的细胞数。在封闭环境上样格式(图1a)中,可以通过选择适当的通道大小和细胞上样浓度来很好地控制高通量。我们展示了使用覆盖约30%的标准1“x3”玻璃载玻片的流量通道每次运行可处理约5000个单个细胞,并且无需对设备设计和工作流程进行重大改动即可轻松实现每次运行可放大至超过15,000个细胞。
为了为scrna-seq应用提供格式灵活性,我们使用以开曲面和封闭环境的上样格式(图1c,1d)的微孔阵列建立了工作流程。在开曲面格式中,微孔阵列被用作微型化的微量滴定板,这种格式具有易于使用的优点,并且具有共同测量来自同一组细胞的mrna和蛋白质的潜力,例如使用完善的单细胞条形码芯片(26)或微雕刻(27)方法。对于scrna-seq,首先使用移液器将细胞和珠子依次上样到阵列上,并通过重力使其沉降到孔中。尺寸排阻确保每个孔最多容纳单个珠子,整个阵列中的珠子上样效率>90%。捕获细胞和珠子后,将高渗溶液(在本文中也称为冻融缓冲液)移液到阵列上,并使用手动夹具用载玻片密封微孔。使用三个冻融循环进行细胞裂解,然后孵育60分钟以使释放的mrna被捕获到条形码珠上(图1b)。孵育后,移出载玻片,并通过离心(阵列放向倒置)取回珠子,用于后续的文库制备步骤,如先前所述(17,23)(图1c,1e)。
在封闭环境格式中,微孔阵列结合到微流体通道上,使用层流图谱引入细胞和试剂(图1a)。由于增加的一致性、更高的上样效率、减少的细胞消耗以及最小化条形码珠的浪费,这种格式是特别有利的。与开孔格式相比,细胞和珠子的上样时间也更短,因为细胞和珠子被限制在由微孔顶部的通道几何形状和厚度所限定的浅区域中,这有利于珠子和细胞沉入孔中。而且,细胞和珠子也可以通过逆向流动而在微通道中来回流动以增加捕获概率,这使得这种格式特别适合于低输入量的样品(例如,总共<500个细胞)。这种格式也更有利于在转录本捕获之前对细胞进行干扰测定,这可能对许多研究(例如药物筛选应用)有用。在没有外围流体处理设备的情况下,我们可以使用重力驱动的流将细胞和珠子依次上样到该设备中,然后通过重力将其沉降到孔中(数据未显示)。由于层流图谱和来回移动珠子的能力,我们通常在封闭环境下达到>99%的珠上样效率。然后将高渗缓冲液引入通道中,然后用氟化油密封。由于缓冲液本身不会引发裂解,因此可以使用重力驱动的流手动进行油封,而没有时间限制,也无需担心材料损失。之后,类似于开曲面格式(图1d,1e)所描述的,进行包括细胞裂解、mrna捕获、珠回收、逆转录、扩增和文库制备在内的下游过程。
两种格式都可以以高度受控的方式进行操作,而不需要任何外围流体处理设备。而且,scftd-seq工作流程是模块化的,这允许在细胞/珠上样后存储样品并在选定的步骤中暂停,而不会降低数据质量,这对于分布式采样和广泛应用是有利的。例如,人们可以使用任一设备来共分离细胞/珠子并冻结下来,以运送回集中式设备进行下游处理,以确保数据质量和一致性(图1e)。这样,scftd-seq结合了市售的基于微孔的scrna-seq平台的最理想方面,并提供了更加用户友好的工作流程减少了动手时间,降低了成本,并且不需要自动化系统或半透膜。
实施例2.技术验证和基准测试
重复的冻融循环是裂解细菌细胞的一种古老方法,并且已被证明可用于哺乳动物细胞,包括整合到微流体系统中进行基因表达分析(28)。这种方法在冷冻过程中导致冰晶形成,解冻后细胞膨胀,最终导致细胞膜破裂。但是,众所周知,反复冻融会破坏大型生物分子并导致断裂,这是全长mrna测序等应用所面临的问题。尽管以前已将其用于促进微孔中基于去污剂的细胞裂解方法(24),但单独的冻融循环(不使用基于裂解/去污剂的缓冲液)尚未被认为是细胞裂解制备单细胞mrna测序文库的可行方法。
我们出乎意料地发现,在基于分子条形码的单细胞3'mrna谱分析的背景下,冻融裂解不会断裂谱分析所需的mrna。不受理论的束缚,这很可能是因为基于分子条形码的单细胞3'mrna谱在3'末端仅需要约50个碱基。令人惊讶的是,以下数据显示,单独的冻融裂解(不使用基于去污剂的裂解缓冲液)可有效裂解细胞,并且细胞释放出足够量的mrna,可用于单细胞3'mrna转录组分析。
我们首先通过对上样到微孔阵列上的gfp表达细胞进行冻融循环并成像细胞裂解液的荧光来直观地确认细胞裂解。即使经过一个冻融循环,我们仍观察到细胞裂解液在整个孔中均匀扩散,这证明了细胞膜的破裂以及大分子(gfp)从破裂的膜中扩散出来的能力(数据未显示)。为了确认核膜的破裂,我们在细胞核中表达了与sv40核定位序列融合的rfp,并且同样观察到即使经过一个单一的冻融循环,细胞质和核裂解物均匀地分布在孔内(数据未显示)。此外,我们观察到裂解后长达2h的荧光强度几乎没有变化,这表明有效的微孔密封和裂解物在微孔中的保留(数据未显示)。
接下来,我们检查了冻融循环是否可以从细胞中释放出与基于去污剂的技术相当的rna量。为此,我们将冻融裂解法与最初在dropseq方法中使用的基于去污剂的方法进行了比较(17)。尽管我们使用基于去污剂的方法检测到的rna量高于冻融循环,尤其是与仅一两个循环相比,但与三个循环相比,差异不大,其中三个循环的产量>以去污剂法为标准的rna含量的90-95%(数据未显示)。然后输入相同量的rna进行定量pcr,我们为人(k562,数据未显示)和小鼠细胞(nih3t3,数据未显示)检测到一组选定基因的相似基因表达水平,证实了冻融裂解法用于定量基因表达研究的可行性,其结果与基于去污剂的技术相当。
为了验证整个scftd-seq工作流程及其技术性能,我们使用开曲面和封闭环境上样格式平台,分析了人类白血病细胞系k562和小鼠成纤维细胞系nih3t3的细胞混合物。大多数细胞条形码与人或小鼠的转录组进行唯一比对,表明物种高度特异性的单细胞谱,双峰频率低,孔间交叉污染最小(图2a,2b)。每个细胞平均读取20万次,我们从人类细胞中的约6000个基因中平均检测到约45,000个转录本,从小鼠细胞中的约5500个基因中检测到约30,000个转录本,表明转录本的捕获效率与上述平台中报道的非常相似(图2c-2f)。读取深度的下采样类似地产生了与其他平台获得的基因和转录物数量相当的基因和转录本数量(图2c,2d)。测序结果还揭示了高质量文库,其中映射定位到外显子区域的读序的平均分数>最小映射定位到核糖体rna区域和线粒体基因的预期表达百分比(<5%,图2g-2i)的88%。所有这些指标均可与包括dropseq(17)、seqwell(23)、indrop(18)和10xgenomics(20)在内的最新单细胞3'mrna测序平台相媲美。
为了证明scftd-seq能够一次处理约5000个单细胞转录组并鉴定异质样品中的细胞类型的能力,我们使用了以1:10比例混合的人(k562)和小鼠(nih3t3)细胞的混合物,并将其上样到最大的scftd-seq平台(70,000孔)上。在比对和过滤后,我们获得了5062个单细胞转录组谱(551个k562细胞和4511个nih3t3细胞),接近预期的1:10混合比例(1:8.19,图3a)。还使用三种人类细胞系k562、hek和huvec以1:1:2的比例混合进行了类似的实验。过滤后,我们恢复了3927个单细胞数据点,并且非监督式的聚类将细胞分为三个主要的集群,每个集群可以根据细胞类型特异性标记的表达推断为相应的细胞类型(图3b,3c)。我们确定了984个k562细胞、1000个hek细胞和1943个huvec细胞,这与预期的1:1:2的混合比例非常吻合。
实施例3.整个肿瘤的单细胞rna分析
为了进一步证明scftd-seq可与新鲜分离的原代细胞一起使用并实现亚群探究,我们对在yummer1.7同系小鼠模型中产生的黑色素瘤肿瘤中解离的细胞进行了分析,其中肿瘤细胞也经过工程改造以表达gfp进行鉴定(29)。过滤后,我们获得了642个细胞,并在每个细胞约25k读取深度处平均检测到约1400个基因和每个细胞约4250个转录本。使用聚类分析,我们分别基于ptprc(cd45)和gfp基因表达鉴定了四个集群,它们形成了两个截然不同的主要集群,被推断为免疫细胞和肿瘤细胞(图4a-4c)。差异基因表达分析表明,免疫细胞簇之一(集群3)富含细胞毒性相关基因的表达(gzmb,gzmc,图4d)。对典型的淋巴细胞标志物(cd3g、cd8、nkg7、trbc1和ctla2a)的检查进一步表明,该集群主要由细胞毒性t细胞和自然杀伤(nk)细胞组成,而其他免疫细胞集群(集群1)包括大量的单核细胞/巨噬细胞(itgam/cd11b、cd14、cd68)和少量树突状细胞(itgax/cd11c)、t细胞(cd3g)和nk细胞(nkg7;数据未显示)。对于肿瘤细胞,我们的分析类似的鉴定出一小部分细胞(集群4),其中富含与调控脉管系统发育和血管生成相关的几个基因(anxa3、ctsh、ccl11、cd34、sfrp2、rap1a;表s2),这表明可能呈现出肿瘤细胞的生物学新亚群。
总体而言,这些结果证明了冻融循环作为测量单细胞基因表达的合适裂解方法的可行性,并进一步确立了scftd-seq作为单细胞3'mrna测序的可靠方法,具有与最新平台相匹配的技术性能。
实施例4.来自sle患者的致病性t细胞的单细胞rna谱分析
辅助性滤泡t细胞(tfh)是在次级淋巴器官(slo)中发现的cd4+t细胞的亚群,可为b细胞分化为浆细胞和记忆细胞提供必要的帮助(30)。动物模型和患者的证据表明,tfh细胞的异常功能与系统性红斑狼疮(sle)中的致病性自身抗体的产生有关,其中sle是一种以免疫复合物介导的多个器官组织损伤为特征的自身免疫疾病(30)。但是,由于对slo样品的获得有限,因此对sle中的tfh细胞的研究一直具有挑战性。过去十年的研究发现,人类血液cd4+cxcr5+细胞包括cd4+t细胞的记忆区室,其与slo中的tfh细胞共享表型和功能特性,从而为分析sle中tfh细胞提供了一个窗口(13,31)。进一步的分析表明,循环性的cd4+cxcr5+t细胞由具有不同表型和功能的不同群体组成,尽管在实验室之间选择用来定义这些血液tfh亚型的标志物组合也有所不同(13,31)。在我们最近的工作中,我们使用pd-1和ccr7标志物将cd4+cxcr5+群体分为两个亚群,并显示cd4+cxcr5+pd-1高ccr7低群体(称为循环性tfh样细胞(ctfh))在sle患者中扩增,与cd4+cxcr5+pd1低ccr7高中枢记忆t(tcm)细胞相比,具有类似于slo中tfh细胞的功能,其特征在于pd-1的表达更高,并且分泌il-21(32)。在这里,我们扩展了我们的早期工作,应用scftd-seq平台表征了这两个细胞群体,以深入了解其转录程序中的异质性。另外,这些实验使我们能够评估scftd-seq与低输入临床样品(例如几千个细胞)的相容性。
在对cd4+cxcr5+pd1高ccr7低和cd4+cxcr5+pd1低ccr7高细胞群进行分类之后(从现在开始,分别称为ctfh和tcm细胞,未显示),我们将每个样品分成两等份;第一份用离子霉素和佛波醇12-肉豆蔻酸13-乙酸酯(pma)激活,而第二份则保留为未经处理的对照。离子霉素/pma刺激触发t细胞活化途径,特别是细胞因子产生的途径,并允许探测广泛的反应以进行功能表征。在这项研究中使用了一个小室设备,并为scftd-seq捕获了约1000个单细胞。所有这些样品都可以在分选后立即上样,但可以存储所需的时间以进行批处理以生成测序库。在这里,我们首先分析每个t细胞组,以寻找不同的亚群。对于未经处理的样品,尽管细胞之间存在相当大的差异,但非监督式的聚类未发现任何明显可区分的聚类(数据未显示)。对于刺激条件,聚类确定了ctfh和tcm细胞的两个主要亚群(数据未显示)。检查差异表达的基因后,我们发现一个亚群显示出更强的激活作用,这可以从细胞因子基因的表达以及趋化因子受体基因cxcr4和ccr7的下调中推断出来,想必这使细胞无法重新进入b细胞滤泡-一种激活的表型-而另一表型中的响应不如从更高的cxcr4和ccr7表达中推断出的响应(它们重新进入卵泡的可能能力),表明这些细胞群中对均匀刺激(离子霉素/pma)的响应水平也有所不同(数据未显示)。
接下来,我们通过非监督式聚类和差异基因表达分析比较了受刺激组和对照组。对于ctfh和tcm组,我们确定了两个主要的集群,它们与受刺激组和对照组基本吻合(图5a,5d)。差异基因表达分析显示,受刺激组中与免疫细胞信号传导和激活相关的基因(尤其是细胞因子基因)上调,而t细胞受体基因(cd3g,cd247)和某些趋化因子受体基因(cxcr4,ccr7)下调(图5b,5c,5e,5f,数据未显示),而这两组基因的下调表示激活。类似地,基因组富集和途径分析显示与两种刺激的t细胞类型的增殖、移动、细胞因子调节和炎性途径相关的基因组的富集(数据未显示)。值得注意的是,虽然刺激能诱导大多数细胞产生强大的细胞因子,但我们还观察到了小部分未经处理的细胞(包括ccl5和ccl20)的趋化因子基因表达(分别编码rantes和mip-3β的基因),提示t细胞具有sle患者血液中循环的活化的炎性表型(图5c,5f)。然而,在刺激后,细胞在细胞因子基因反应中表现出显著的多功能性,因为我们观察到>60%的细胞具有这种能力(数据未显示)。
然后,我们一起分析了受刺激的ctfh和tcm组,以揭示其效应子功能的任何相似之处和不同之处。我们首先进行了t分布随机邻接嵌入(t-sne)分析,其中具有相似转录谱的细胞聚集在一起。当我们在t-sne图上检查ctfh和tcm细胞的分布时,我们注意到两种细胞类型在第一个tsne组分上是可分离的,其中ctfh细胞主要向右聚集,而大多数tcm细胞仍留在左侧(图5g)。在主成分分析中也可以观察到这种模式,并且在第一主成分中这两种细胞类型尤其可分离,所属第一主成分由选定的细胞因子和趋化因子基因组成,包括il2、tnf、ifng和ccl20(mip3a)(数据未显示)。与这些视觉观察一致,非监督式聚类将细胞分为三组,其中集群1主要由tcm细胞组成,集群2和3主要由ctfh细胞组成(数据未显示)。我们还通过差异基因表达分析捕获了这些差异,并发现,尽管两种活化的细胞类型均显示出细胞因子基因的急剧上调,但它们的细胞因子谱却有很大差异(图5h,5i,未显示数据)。尤其是,尽管tcm细胞大量表达il8、il23a、tnf和ccl20,但ctfh细胞却几乎只表达il4、il21、ccl3(mip1α)、ccl4(mip1β)、ccl5(rantes),并表现出更高的ifng和il10的表达(数据未显示)。这些结果表明,ctfh细胞是具有炎症功能的异质效应细胞群体,并且与tcm细胞不同。我们还观察到,il2表达在两种细胞类型中均遵循双峰分布,且表达水平相当(数据未显示)。与健康受试者相比,由于来自sle的循环t细胞产生的il2转录本和il-2蛋白产量降低,并伴随着t细胞受体介导的激活功能改变(33,34),因此有必要对这两个不同的种群进行进一步比较。与刺激条件相似,我们还观察到未经处理的ctfh和tcm细胞之间的基线基因表达水平存在差异(数据未显示)。
总体而言,这些结果描绘了单细胞水平上ctfh和tcm细胞的转录谱,并显示了在刺激时和在基线水平上,其效应子功能的内在差异。cc趋化因子家族(包括ccl3、ccl4和ccl5)在ctfh细胞中的特异性表达表明它们通过促进炎症反应并可能激活炎症白细胞而在炎症中潜在的致病作用。
我们推出了scftd-seq,这是一种基于冻融裂解的单细胞3'mrna-seq方法,适用于微孔阵列平台。我们通过几个关键的演示验证了scftd-seq的技术性能:(1)使用包括先前sc-rnaseq出版物中用于头对头比较的细胞系和混合样本来区分异型群体的能力,(2)使用原代细胞,包括来自整个肿瘤样品的所有类型的细胞和来自患者的罕见t细胞群,以及(3)检查刺激后t细胞的活化状态。我们已经建立了具有与先进平台可比的技术性能(通量、转录本捕获效率、单细胞分辨率、低输入样品兼容性)的常规实践。使用scftd-seq条形码化单细胞来源的mrna的成本(每个样品约150美元,主要是珠子成本和nexteraxt试剂盒)比商业平台(例如10倍铬(每个样品>1500美元))低一个数量级。与以前建立的技术相比,我们的平台还具有一些关键优势。首先,冻融裂解消除了任何复杂的准备/后续步骤和任何操作难题,从而减少了方案步骤的数量并简化了总体操作程序。其次,我们的平台提供了格式灵活性,其将以最适合实验需求的配置实现scrna-seq应用。重要的是,两种格式都可以手动操作,而无需任何其他外围设备,从而便于携带和使用。第三,工作流程是模块化的,可以在细胞-珠共分离步骤暂停,而对数据质量没有影响,从而减少了连续操作细胞分离、裂解和反转录所产生的麻烦,因此更适合于应用在小型诊所或即时医疗点等分布式站点。这样,scftd-seq提供了一个可靠、高效的平台,具有简化的模块化工作流程,可在学术、临床和大规模服务环境中更轻松地被采用。
我们通过将scftd-seq应用于从sle患者获得的循环性人cd4+cxcr5+t细胞,证明了其实用性。此类细胞主要具有中枢记忆表型,具有cxcr5和ccr7表达赋予的能力,可迁移至b细胞滤泡以引发继发性t依赖性b细胞应答(35)。cxcr5+池中还包含具有激活表型的细胞,pd-1上调而ccr7下调,即所谓的循环性tfh细胞,这些细胞可能会返回cxcr5阳性记忆池(36)。我们和其他人已经表明,被鉴定为cd4+cxcr5hipd1高的这一亚群在sle患者的血液中扩增(32,36-38)。这些细胞产生il-21,其pd-1表达与疾病活动相关。与循环性cxcr5+中枢记忆细胞群相比,它们表达的ccr7量更低,从而能够区分流式细胞术(32,36)。
与cd4+cxcr5+pd1低ccr7高(tcm)细胞相比,我们在这里扩展了我们的早期工作并探索了cd4+cxcr5+pd1高ccr7低t细胞(ctfh)的单细胞转录情况,从而可以观察其异质性和效应子功能。scrna-seq可以识别具有激活特征(细胞因子基因和cd69的表达)的细胞,即使在未经处理的对照组群体中,由于单细胞分辨率的影响,其数量仍然很少。虽然通常会执行刺激策略来评估细胞功能,但通过scftd-seq技术灵敏地检测少量活化细胞的能力可以评估功能状态而无需刺激,即使在基线水平也是如此。另一个观察结果是,在两个细胞组中观察到,受刺激细胞群产生多种促炎细胞因子转录本的能力(如果表达的话),可能会导致sle中常见的炎症状态(39)。这样的多功能性也可能有助于疾病活动,值得进一步研究。最后,我们观察到,尽管两个细胞群均显示出强劲的细胞因子基因表达,但它们的细胞因子谱与主要表达ifng、il4、il21、mip1α、mip1β和rantes的ctfh细胞截然不同-炎症性细胞因子转录本可能有助于促进自身反应性b细胞活化并引起炎性组织损伤。总而言之,这些观察为ctfh和tcm细胞的功能状态提供了新的见解,可能有助于解释它们在sle中的作用。比较患者和健康受试者之间的细胞谱可确保阐明sle发病机理,并确定潜在的生物标志物和治疗靶标。
材料与方法
微孔阵列制造和设备组装
使用su-8负性抗蚀剂制造用于微孔阵列的主晶片。将单层抗蚀剂(su-82035,microchem)以2200-2400rpm旋转30秒,以得到约50μm的特征高度。然后通过透明蒙版(cad/艺术服务)将晶片暴露于紫外线下以对微孔进行构图。显影和烘烤后,将晶片在150℃硬烘烤30分钟,并在用三氯甲基硅烷(sigma-aldrich)饱和的真空室中硅烷化2小时。在封闭格式操作中使用的微流体通道的制造遵循类似的制造程序,使用su-82075,其中通道高度设置为约120μm(1700-1800rpm,持续30s)。
通过在主晶片上浇铸聚二甲基硅氧烷(pdms,sylgard184,道康宁公司),然后在80℃下脱气和固化6-8小时来制造设备。对于封闭格式,将微孔阵列和微流体通道设备的最终高度都设置为3-4mm。对于开放格式,通过旋涂pdms将微孔阵列的高度设置为<0.5mm,以使该阵列与载玻片结合时与安捷伦夹具(安捷伦,g2534a)兼容。固化后,将pdms剥离,然后将设备切成合适的尺寸以适合载玻片。对于微流体通道,使用活检打孔器(miltex,1.5mm)刺破用于流体连接的孔。对于开放格式,将微孔阵列等离子键合到载玻片上。对于封闭格式,首先将微孔阵列等离子键合到微流体通道,然后再键合到载玻片上。
scftd-seq操作–开曲面上样格式
对于开曲面格式,细胞和珠的上样程序主要采用seqwell方法(23),而无需使用纳米多孔膜。在细胞上样之前,将微孔阵列进行等离体暴露以使微孔表面亲水,然后浸入pbs中的1%牛血清白蛋白(bsa)中30分钟以激发。然后用pbs洗涤阵列,并使用移液管除去阵列顶部的溶液,仅留下溶液的薄层。然后将单细胞悬液(在100-200μlpbs+1%bsa溶液中的10,000-15,000个细胞)移液到阵列的多个位置以覆盖整个阵列区域,并通过手动摇动阵列将其间歇孵育10分钟,以提高细胞上样。孵育后,微孔阵列用pbs洗涤以除去未沉降的细胞。重复洗涤多次,以确保去除多余的细胞,并通过成像确认。细胞上样后,将mrna捕获珠(macosko-2011-10,chemgenes)作为150,000个珠以100-150μl的浓度悬浮在pbs中,移液到阵列上,并通过重力上样到微孔中,这与对细胞的描述相似。手动间歇性摇动阵列,以确保>90%的孔的上样量。使用pbs洗涤去除多余的珠子,并通过成像确认。然后将冻融裂解缓冲液(体积为500μl;组成:100mmtris–ph7.5、10mmedta,1mnacl,5μmdtt,0.4u/mllucigenrna酶)移至阵列上,并将其在室温下孵育5分钟。除去过量的冻融裂解缓冲液,在阵列顶部留下约200μl缓冲液。然后小心地使用载玻片密封阵列,并使用手动夹具(安捷伦科技公司,g2534a)固定。进行三个冻融循环以裂解细胞,将细胞在-80℃冰箱或干冰/乙醇浴中冷冻10分钟,然后在室温下融化10分钟。裂解后,将微孔阵列在湿室内孵育1小时,以将mrna捕获到珠上。孵育后,将微孔阵列松开,小心地移出载玻片。然后将阵列与边载玻片对接,然后以反向定向转移到充满pbs的4孔板中,与seqwell方法(23)中所述的类似。将4孔板以1000g离心5分钟以将珠释放到pbs溶液中。通过用载玻片刮擦阵列表面,小心除去所有剩余的珠子。将含有珠子的pbs溶液转移到15ml离心管中,以1000g离心5分钟,然后转移到1ml体积的1.5ml微量离心管中,进行逆转录。
scftd-seq操作–封闭环境上样格式
对于封闭环境格式,首先将微流体设备注入pbs,然后使用手动操作的注射器将其加压,以去除微孔内部的气泡,并关闭出口。用pbs的1%bsa填充设备,并在室温下孵育30分钟,以防止细胞和分子附着在pdms表面上。然后在装入细胞和珠子之前用pbs清洗设备。为了建立重力驱动的流动,将设备出口连接到10英寸的管道,末端连接一个单路塞,而设备入口保持未连接状态,以用作水库。在这种配置中,只需将溶液吸移到入口储液罐中,然后通过调节入口与连接到出口的管道末端之间的高度差,通过重力驱动的流动将其抽回设备中。单路塞还允许对流体流进行启动/停止控制,以促进细胞和珠的上样。类似地,也可以通过调节管道的高度在出口侧产生更高的液体静压力来逆转流量。在scrna-seq实验过程中,将单细胞悬液(50μlpbs+1%bsa溶液中的5,000-10,000个细胞)吸移到进样口中,然后抽回设备中。一旦通道完全充满细胞溶液,就停止流体流动,并通过重力使细胞沉降。用pbs冲洗掉多余的细胞,并以类似于细胞的方式上样50-150μl中的30,000-120,000个mrna捕获珠。尺寸排阻和来回上样确保单个珠可上样>99%的微孔。用pbs冲洗掉多余的珠子,然后将100-200μl冻融裂解缓冲液引入设备中。然后将体积为100-200μl的氟化油(fluorinertfc-40)抽入设备中以密封微孔。油封后,断开出口处的管,并将微流体装置暴露于三个冻融循环,在-80℃冰箱或干冰/乙醇浴中冷冻5分钟,在室温下融化5分钟。裂解后,将微流体装置在湿室内孵育一小时,以将mrna捕获到珠上。孵育后,将微流控设备的入口连接到装有6x盐水-柠檬酸钠(ssc)缓冲液的注射器上,并将出口连接到带有管子的微量离心管。然后将微流体装置倒置,并通过吹扫将珠子从装置中冲洗出至管中。在吹扫之前以倒置方向离心微流体装置,或在吹扫过程中用镊子轻轻敲打微流体装置的背面,以助于将珠移出微孔。使用这种方式,我们能够回收>95%的珠子。将收集的珠子以1000g离心1分钟,并在逆转录之前用6xssc缓冲液洗涤两次。
文库制备和测序
文库制备和测序步骤遵循与dropseq方法(17)和seqwell方法(23)中相同的步骤,其中详细描述了缓冲液的组成和方案步骤。简而言之,取出后首先用6xssc洗涤珠子两次,然后使用maximahminus逆转录酶(thermofisher)和定制的模板转换寡核苷酸对捕获的mrna进行逆转录。然后在37℃下用核酸外切酶i(exoi,neb)处理cdna包被的珠子45分钟,以去掉任何未结合的mrna捕获探针。然后如seqwell方法(23)所示,使用半pcr反应扩增包被cdna的珠子,对细胞系或大细胞使用13个循环,对原代细胞使用16个循环。使用ampurexp珠(贝克曼库尔特(beckmancoulter))以0.6的比例纯化扩增的dna,并使用高灵敏度芯片通过agilentbioanalyzer评估扩增的dna的质量。然后合并纯化的cdna,并使用定制引物代替i5内参引物输入标准的nextera标签和扩增反应(nexteraxt,illumina),仅扩增那些包含细胞条形码和umi的片段。然后使用ampurexp珠以0.6x的比例纯化pcr产物,并通过agilentbioanalyzer高灵敏度芯片检查文库的质量。使用针对读序1的定制引物在hiseq2500测序仪(illumina)上对文库进行测序,其中读序1具有75个循环,读序2具有75个循环。对于读序1,仅使用前20个碱基进行分析。phix库以20%作为加标对照使用。
数字基因表达矩阵的读序比对和产生
如dropseq方法(23)中所述,使用dropseq工具(http://mccarrolllab.com/dropseq)进行了包括条形码/umi识别和折叠在内的转录组比对。简而言之,将包含转录本信息的第二个读段在5'端和3'端修剪,分别去除衔接子序列和polya尾巴,并在第一个读段中用细胞条形码和umi序列标记。然后使用starv2.5.2b将读序与相应物种(小鼠,mm10;人,hg19;人-小鼠混合物,hg19_mm10)的参考转录组进行比对。唯一比对的读序首先按细胞条形码分组,然后按具有单碱基错误容限的umi分组。每个基因的不同umi序列的总数报告为数字基因表达矩阵中与该基因相对应的转录本数。
单细胞基因表达数据分析
使用seurat(25)(biorxiv:https://doi.org/10.1101/164889)对归一化和对数转换的基因表达数据进行数据分析。对每个细胞进行文库大小归一化,其中每个基因的转录本数量由转录本总数乘以10,000来计算。
目视确认冻融裂解
将表达gfp的huvec细胞(angio-proteomie)在egm-2mv完全培养基(lonza,cc-3202)中于37℃和5%co2下生长,并在>80%汇合的情况下进行胰蛋白酶处理。制备单细胞悬浮液,并如上所述在微孔阵列上样,而没有珠。然后将细胞暴露于以下三个循环中:冻融、在-80℃冰箱或干冰/乙醇浴中冷冻并在室温下解冻。在随机选择的位置拍摄裂解之前、一个冻融循环和三个冻融循环后细胞的代表性图像。通过gfp在整个微孔体积中的分布来证实细胞裂解。为了确认核裂解,使用表达与rf40核定位序列融合的rfp的细胞光核-rfp(thermofisher)转染细胞。温育过夜后,在荧光显微镜下确认核染色,并如上所述重复冻融实验。rfp在整个微孔体积中的分布证实了核溶解。
冻融裂解效率实验
将人k562细胞(atcc)在rpmi培养基中生长,该培养基中添加了10%的热灭活的fbs、2mm的l-谷氨酰胺、0.1mm的2-巯基乙醇、100单位/毫升的青霉素g钠和100μg/ml的硫酸链霉素。将nih3t3小鼠成纤维细胞(atcc)在含有10%小牛血清、4mml-谷氨酰胺和100单位/ml青霉素g钠和100μg/ml硫酸链霉素的dmem培养基中培养。对于k562和nih3t3,均制备了100,000个细胞的单细胞悬液,并使用冻融循环(在冻融裂解缓冲液中;100mmtris–ph7.5、10mmedta、1mnacl、5μmdtt、0.4u/mllucigenrnase中)以500μl体积裂解,或dropseq方法(17)中使用的基于去污剂的缓冲液(100mmtris-ph7.5、10mmedta、3%ficollpm-400、0.1%sarkosyl、25mmdtt)。对于冻融裂解,将裂解液分别进行1、2和3个冻融循环进行比较。裂解后,按照制造商的说明,使用磁性oligo-dt珠(neb,s1419s)从裂解物中分离出mrna。使用nanodrop2000分光光度计对mrna进行定量。对于qpcr,将10ngmrna作为所有条件的输入,并按照制造商的说明,使用ssofastevagreenpcrsupermix(biorad)在cfxconnect实时pcr机(biorad)上进行反应。引物获自sigma(kicqstart引物)。
物种混合实验
如上所述,将相等数量的人k562和小鼠nih3t3细胞混合并上样到微孔阵列上,并在中等深度(平均20,000-40,000个读序/细胞)进行测序。比对后,基于>10,000个读序、>5000个转录本和>1000个基因过滤细胞。与物种特异性转录组的转录本比对,>90%的细胞被鉴定为属于该物种。为了计算转录本捕获效率,对细胞进行了更高深度的测序(平均每细胞200,000个读序),并使用picardtools(broadinstitute.github.io/picard/)对读序进行降采样,以提供每个细胞不同平均读序时的适当比较。
对于以1:10比例进行的物种混合实验,按所述,将k562细胞和nih3t3细胞以1:10比例进行混合,上样到微孔阵列设备上,并以较浅的测序深度(平均5000个读序/细胞)进行测序。根据>1000个读序和>500个转录本过滤细胞。与上述相似,将与物种特异性转录组的转录本比对的>90%的细胞鉴定为属于该物种。
hek-k562-huvec混合实验
将hek、k562和huvec细胞以1:1:2的比例混合,并上样到微孔阵列上。对细胞进行浅层测序(平均3500个读序/细胞),并根据>1500个读序、>1000个转录本和>300个基因进行过滤。使用seurat软件包(25)(biorxiv:https://doi.org/10.1101/164889)进行聚类和差异基因表达分析,并根据细胞类型特异性基因表达的表达推断细胞身份。
黑色素瘤实验
为了对yummer1.7黑色素瘤进行单细胞分析,将0.5x10^6yummer1.7-gfp细胞皮下注射到6周龄雄性c57bl/6j小鼠的腹侧。让肿瘤生长21天;使用串行卡尺测量追踪肿瘤体积。在第21天,当肿瘤的体积大约为500mm3时,对小鼠实施安乐死并收获肿瘤。通过在37℃轻度振荡下,用含有2%fbs的rpmi的1000u/ml的iv型胶原酶(sigma)消化30分钟,生成单细胞肿瘤悬液。然后使用cd45(ebioscience)和live/dead可固定的远红色死细胞染色试剂盒(thermofisher)对细胞染色。通过facs纯化细胞。排除死细胞和碎片,并富集cd45+细胞,以增加用于测序的cd45+细胞的频率,cd45+和cd45-细胞的最终比例为1:1。将细胞分类到含10%fbs的冷rpmi中,并在处理前置于冰上。对于测序实验,将细胞上样到微孔阵列上,并如上所述以每个细胞约25,000个读序的平均深度进行测序。根据>8000个读序、>1000个转录本和>300个基因过滤细胞,并使用seurat软件包(25)(biorxiv:https://doi.org/10.1101/164889)进行分析。
sle患者循环cd4+cxcr5+t细胞实验
通过在ficoll-paque(gehealthcare)上进行密度梯度离心,从新鲜血液中分离外周血单个核细胞(pbmc),并将单细胞悬液用以下抗体染色:apc-h7偶联的抗cd3(克隆sk7)、pe-cy5偶联的抗cd45ra(克隆hi100)、pe偶联的抗cd25(克隆m-a251)、pe-cy7偶联的抗pd-1(克隆eh12.1)、alexafluor488偶联的抗cxcr5(克隆rf8b2)和v450偶联的抗ccr7(克隆150503)(全部来自bdbioscience),和alexafluor700偶联的抗cd4(克隆okt4;来自ebioscience)。通过facsaria(bdbiosciences)将染色的细胞分类为原初(cd3+cd4+cd45ra+ccr7hipd-1lo)、cxcr5+中枢记忆(tcm:cd3+cd4+cd45ra-cxcr5+cd25loccr7高pd-1低)和循环tfh(ctfh:cd3+cd4+cd45ra-cxcr5hicd25loccr7低pd-1高),排除了前向和侧向散射造成的双峰。
对于测序实验,将细胞上样到微孔阵列上,并如上所述以每个细胞约20,000-40,000读序的平均深度进行测序。基于>10000个读序、>1000个转录本和>400个基因过滤细胞,并使用seurat软件包(25)(biorxiv:doi.org/10.1101/164889)进行分析。
参考文献
1.tang,f.,lao,k.和surani,m.a.(2011)单细胞转录组分析的开发与应用.natmeth.
2.eberwine,j.,sul,j.-y.,bartfai,t.和kim,j.(2014)单细胞测序的希望.natmeth,11,25-27.
3.jaitin,d.a.,kenigsberg,e.,keren-shaul,h.,elefant,n.,paul,f.,zaretsky,i.,mildner,a.,cohen,n.,jung,s.,tanay,a.等人(2014)用于组织的无标记分解成细胞类型的大规模平行单细胞rna-seq.science,343,776-779.
4.
5.gaublomme,jellertt.,yosef,n.,lee,y.,gertner,ronas.,yang,liv.,wu,c.,pandolfi,pierp.,mak,t.,satija,r.,shalek,alexk.等人(2015)单细胞基因组学揭示了th17细胞致病性的关键调控因子.cell,163,1400-1412.
6.shalek,a.k.,satija,r.,adiconis,x.,gertner,r.s.,gaublomme,j.t.,raychowdhury,r.,schwartz,s.,yosef,n.,malboeuf,c.,lu,d.等人(2013)单细胞转录组学揭示免疫细胞中表达和剪接的双峰性.nature,498,236-240.
7.zeisel,a.,manchado,a.b.m.,codeluppi,s.,
8.marques,s.,zeisel,a.,codeluppi,s.,vanbruggen,d.,mendanha
9.papalexi,e.andsatija,r.(2017)单细胞rna测序探索免疫细胞异质性.natrevimmunol,提前在线出版.
10.bendall,seanc.,davis,karal.,amir,e.-add.,tadmor,michelled.,simonds,erinf.,chen,tiffanyj.,shenfeld,danielk.,nolan,garryp.andpe’er,d.(2014)单细胞轨迹检测发现人类b细胞发育的进展和调节协调.cell,157,714-725.
11.drissen,r.,buza-vidas,n.,woll,p.,thongjuea,s.,gambardella,a.,giustacchini,a.,mancini,e.,zriwil,a.,lutteropp,m.,grover,a.等人.(2016)通过单细胞rna测序鉴定不同的骨髓祖细胞-分化途径.17,666.
12.schlitzer,a.,sivakamasundari,v.,chen,j.,sumatoh,h.r.b.,schreuder,j.,lum,j.,malleret,b.,zhang,s.,larbi,a.,zolezzi,f.等人.(2015)cdc1和cdc2呈递的dc祖细胞的鉴定揭示了骨髓中常见dc祖细胞阶段的早期谱系启动.16,718.
13.schmitt,n.,bentebibel,s.-e.andueno,h.人血中记忆tfh细胞的表型和功能.trendsinimmunology,35,436-442.
14.trapnell,c.,cacchiarelli,d.,grimsby,j.,pokharel,p.,li,s.,morse,m.,lennon,n.j.,livak,k.j.,mikkelsen,t.s.andrinn,j.l.(2014)细胞命运决定的动力学和调节因素.natbiotech,32,381-386.
15.islam,s.,zeisel,a.,joost,s.,lamanno,g.,zajac,p.,kasper,m.,lonnerberg,p.andlinnarsson,s.(2014)具有独特分子识别物的定量单细胞rna-seq.natmeth,11,163-166.
16.fan,h.c.,fu,g.k.andfodor,s.p.a.(2015)单细胞的组合标记以进行基因表达细胞计数.science,347.
17.macosko,evanz.,basu,a.,satija,r.,nemesh,j.,shekhar,k.,goldman,m.,tirosh,i.,bialas,allisonr.,kamitaki,n.,martersteck,emilym.等人.(2015)使用纳升液滴对单个细胞进行高度并行的全基因组表达谱分析.cell,161,1202-1214.
18.klein,allonm.,mazutis,l.,akartuna,i.,tallapragada,n.,veres,a.,li,v.,peshkin,l.,weitz,davida.andkirschner,marcw.(2015)用于胚胎干细胞的单细胞转录组学的液滴条形码.cell,161,1187-1201.
19.goldstein,l.d.,chen,y.-j.j.,dunne,j.,mir,a.,hubschle,h.,guillory,j.,yuan,w.,zhang,j.,stinson,j.,jaiswal,b.等人.(2017)大规模并行的基于纳诺韦尔的单细胞基因表达谱.bmcgenomics,18,519.
20.zheng,g.x.y.,terry,j.m.,belgrader,p.,ryvkin,p.,bent,z.w.,wilson,r.,ziraldo,s.b.,wheeler,t.d.,mcdermott,g.p.,zhu,j.等人.(2017)单个细胞的大规模并行数字转录谱.8,14049.
21.hochgerner,h.,
22.yuan,j.andsims,p.a.(2016)用于大规模单细胞rna-seq的自动化微孔平台.6,33883.
23.gierahn,t.m.,wadsworthii,m.h.,hughes,t.k.,bryson,b.d.,butler,a.,satija,r.,fortune,s.,love,j.c.和shalek,a.k.(2017)seq-well:以高通量对单个细胞进行便携式低成本rna测序.natmeth,14,395-398.
24.bose,s.,wan,z.,carr,a.,rizvi,a.h.,vieira,g.,pe’er,d.和sims,p.a.(2015)用于单细胞rna印刷和测序的可扩展的微流控技术.genomebiology,16,120.
25.satija,r.,farrell,j.a.,gennert,d.,schier,a.f.和regev,a.(2015)单细胞基因表达数据的空间重建.natbiotech,33,495-502.
26.lu,y.,xue,q.,eisele,m.r.,sulistijo,e.s.,brower,k.,han,l.,amir,e.-a.d.,pe’er,d.,miller-jensen,k.和fan,r.(2015)单细胞效应子功能的高度多重分析揭示了对致病性配体的深层功能异质性.美国国家科学院院刊,112,e607-e615.
27.love,j.c.,ronan,j.l.,grotenbreg,g.m.,vanderveen,a.g.和ploegh,h.l.(2006)用于快速选择产生抗原特异性抗体的单细胞的微雕刻方法.24,703.
28.toriello,n.m.,douglas,e.s.,thaitrong,n.,hsiao,s.c.,francis,m.b.,bertozzi,c.r.和mathies,r.a.(2008)用于单细胞基因表达分析的集成微流生物处理器.美国国家科学院院刊,105,20173-20178.
29.wang,j.,perry,c.j.,meeth,k.,thakral,d.,damsky,w.,micevic,g.,kaech,s.,blenman,k.和bosenberg,m.(2017)紫外线诱导的体细胞突变在yummer1.7小鼠黑素瘤模型中引发功能性t细胞应答.色素细胞与黑色素瘤研究,30,428-435.
30.craft,j.e.(2012)卵泡辅助性t细胞的免疫和全身性自身免疫.8,337.
31.ueno,h.,banchereau,j.和vinuesa,c.g.(2015)人和小鼠t卵泡辅助细胞的病理生理学.16,142.
32.choi,j.-y.,ho,j.h.-e.,pasoto,s.g.,bunin,v.,kim,s.t.,carrasco,s.,borba,e.f.,
33.kyttaris,v.c.,wang,y.,juang,y.-t.,weinstein,a.和tsokos,g.c.(2007)系统性红斑狼疮患者t细胞中升高的nf-atc2水平差异调节cd154和il-2基因.免疫学杂志,178,1960-1966.
34.linker-israeli,m.,bakke,a.c.,kitridou,r.c.,gendler,s.,gillis,s.和horwitz,d.a.(1983)系统性红斑狼疮(sle)患者白细胞介素1和白细胞介素2的产生缺陷.免疫学杂志,130,2651-2655.
35.macleod,m.k.l.,david,a.,mckee,a.s.,crawford,f.,kappler,j.w.和marrack,p.(2011)表达cxcr5的记忆cd4t细胞为b细胞提供加速的帮助.免疫学杂志,186,2889-2896.
36.he,j.,tsai,louism.,leong,yewa.,hu,x.,ma,cindys.,chevalier,n.,sun,x.,vandenberg,k.,rockman,s.,ding,y.等人.(2013)循环前体ccr7lopd-1hicxcr5+cd4+t细胞表明tfh细胞活性并促进抗原再暴露后的抗体反应.免疫,39,770-781.
37.morita,r.,schmitt,n.,bentebibel,s.-e.,ranganathan,r.,bourdery,l.,zurawski,g.,foucat,e.,dullaers,m.,oh,s.,sabzghabaei,n.等人.(2011)人血cxcr5+cd4+t细胞是t滤泡细胞的组成部分,并含有差异性支持抗体分泌的特定亚集.免疫,34,108-121.
38.simpson,n.,gatenby,p.a.,wilson,a.,malik,s.,fulcher,d.a.,tangye,s.g.,manku,h.,vyse,t.j.,roncador,g.,huttley,g.a.等人.(2010)类似于卵泡辅助性t细胞的循环性t细胞的扩增是一种固定的表型,可识别出严重的系统性红斑狼疮的一个亚型.关节炎和风湿病,62,234-244.
39.lu,r.,munroe,m.e.,guthridge,j.m.,bean,k.m.,fife,d.a.,chen,h.,slight-webb,s.r.,keith,m.p.,harley,j.b.和james,j.a.(2016)先天性和适应性血清介质的失调先于系统性红斑狼疮分类,并提高了自身抗体的预后准确性.自身免疫杂志,74,182-193.
本文所公开的所有参考文献、专利和专利申请相对于其所引用的主题通过引用并入本文,在某些情况下,其可能包含整个文档。
除非明确相反地指出,否则本文中在说明书和权利要求书中使用的不定冠词“一个”和“一种”应理解为表示“至少一个”。
还应该理解的是,除非有明显的相反指示,否则在本文要求保护的包括一个以上步骤或动作的任何方法中,方法的步骤或动作的顺序不一定限于所引用的方法的这些步骤或动作的顺序。
在权利要求书以及以上说明书中,所有过渡短语例如“包括”、“包含”、“携带”、“具有”、“含有”、“涉及”、“持有”、“由……组成”等等应理解为开放式的,即意指包括但不限于。如美国专利局专利审查程序手册第2111.03节所述,仅过渡短语“由……组成”和“基本上由……构成”应分别是封闭的或半封闭的过渡短语。
数值之前的术语“大约”和“基本上”是指所述数值的±10%。
在提供值的范围的情况下,在此特别考虑和描述了该范围的上端和下端之间的每个值。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



