
 商标分类
商标分类  商标转让
商标转让 
五种来源于烈味脚骨脆的克罗烷型二萜类化合物及其制备方法和用途与流程
 2021-02-02 16:02:45|
2021-02-02 16:02:45| 385|
385| 起点商标网
起点商标网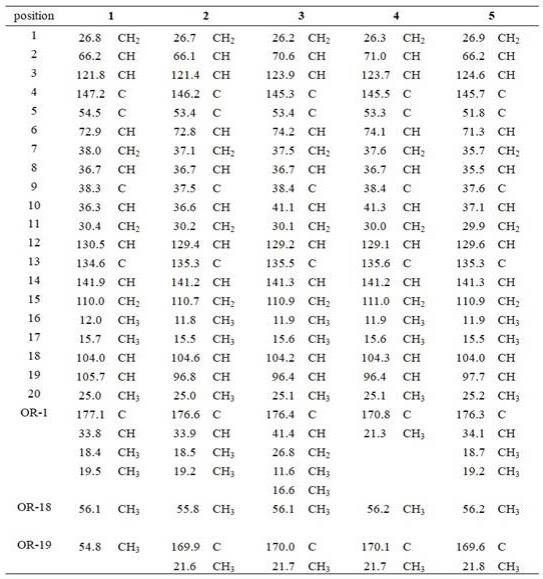
[0001]
本发明属于医药技术领域,具体涉及烈味脚骨脆中新的克罗烷型二萜类化合物及其制备方法和应用。
背景技术:
[0002]
细胞增殖是细胞生命活动的重要特征之一,肿瘤细胞具有不同于正常细胞的增殖特点,主要表现为无限增殖,即永生化。肿瘤细胞永生使细胞周期发生紊乱,细胞的增殖体系不受控制。细胞的增殖依赖于细胞分裂,一个完整的细胞周期包括四个时相,即g1期、s期、g2期和m期。药物作用于细胞周期的任何一个阶段都可以阻断细胞的持续增殖,诱导其走向凋亡。
[0003]
天然产物为我们提供了大量的先导化合物用于进一步的新药开发,如青蒿素用于抗疟,长春新碱用于抗癌等。目前临床上所使用的抗癌药物,大多是干扰或阻断细胞的增殖过程,一般称为细胞毒药物。基于此,我们建立了人肺癌细胞系a549和人肝癌细胞系hepg2细胞筛选模型。在细胞指数生长期加入不同浓度的受试药物,处理一定时间后加入mtt测定光密度值来评判化合物细胞毒性,从而筛选出有较强细胞毒活性的化合物并确定对其最敏感的细胞株,最终发现具有细胞毒活性的化合物,为其进一步用于抗癌药物的开发提供基础。
技术实现要素:
[0004]
本发明的目的在于提供烈味脚骨脆中5个新的克罗烷型二萜类化合物及其制备方法和应用。
[0005]
本发明提供的新化合物1-5属于克罗烷型二萜,其结构如图1所示。
[0006]
本发明还提供了所述新化合物1-5的制备方法,该方法包括如下步骤:(1)烈味脚骨脆(casearia graveolens dalzell)的叶用溶剂提取,回收提取液得粗提物;(2)步骤(1)所得粗提物溶解于水中,采用与水不相混溶的有机溶剂萃取,回收溶剂得到萃取物;(3)步骤(2)所得萃取物经硅胶柱色谱法分离,以石油醚/丙酮或石油醚/乙酸乙酯混合溶剂梯度洗脱;(4)上述步骤(3)中所得流份经mplc(中压液相色谱, 色谱填料为ods)分离,以甲醇/水,或乙腈/水混合溶剂为流动相梯度洗脱;(5)上述步骤(4)中所得流份经hplc-ri(高效液相-示差检测)色谱分离,以甲醇/水为流动相洗脱,或以乙腈/水为流动相梯度洗脱,得到化合物1-5。
[0007]
本发明提供的新化合物1-5的制备方法,所述烈味脚骨脆为大风子科(flacourtiaceae)脚骨脆属烈味脚骨脆(casearia graveolens dalzell)茎叶的提取物。
[0008]
本发明提供的新化合物1-5的制备方法,步骤(1)所述的提取方法为加热回流提取或超声提取,所用溶剂为二氯甲烷、氯仿、乙酸乙酯、甲醇、乙醇中的至少一种,药材:溶剂的重量体积比为1: 5~1: 15。
[0009]
本发明提供的新化合物1-5的制备方法,步骤(2)所述的萃取方法,所用有机溶剂为石油醚、二氯甲烷、氯仿、乙酸乙酯中的任意一种,水溶液和有机溶剂体积比1: 1~1: 2。
[0010]
本发明提供的新化合物1-5的制备方法,步骤(3)中,洗脱溶剂为石油醚/丙酮或石油醚/乙酸乙酯混合溶剂,其比例为100: 2~100: 30。
[0011]
本发明提供的新化合物1-5的制备方法,步骤(4)中,所述甲醇/水混合溶剂的比例为6: 4~9: 1,优选7: 3~8: 2,或乙腈/水混合溶剂比例为6: 4~8: 1,优选7: 3~8: 2。
[0012]
本发明提供的新化合物1-5的制备方法,步骤(5)中所述流动相甲醇和水混合溶剂、或乙腈和水混合溶剂的体积比例为6: 4~9: 1,优选7: 3~9: 1。
[0013]
本发明提供的五个新的萜类化合物对人肺癌细胞a549及人肝癌细胞hepg2具有细胞毒活性。用于制备抗癌药物。
附图说明
[0014]
图1 本发明化合物1-5的结构式;图2 本发明化合物1的
1
h nmr谱;图3 本发明化合物1的
13
c nmr谱;图4 本发明化物1的dept135谱;图5 本发明化合物1的hmqc谱;图6 本发明化合物1的hmbc谱;图7 本发明化合物1的
1
h-1
h cosy谱;图8 本发明化合物2的
1
h nmr谱;图9 本发明化合物2的
13
c nmr谱;图10 本发明化合物2的hmqc谱;图11 本发明化合物2的hmbc谱;图12 本发明化合物3的
1
h nmr谱;图13 本发明化合物3的
13
c nmr谱;图14 本发明化合物3的hmqc谱;图15 本发明化合物3的hmbc谱;图16 本发明化合物4的
1
h nmr谱;图17 本发明化合物4的
13
c nmr谱;图18 本发明化合物4的hmqc谱;图19 本发明化合物4的hmbc谱;图20 本发明化合物5的
1
h nmr谱;图21 本发明化合物5的
13
c nmr谱;图22 本发明化合物5的hmqc谱;图23 本发明化合物5的hmbc谱;图24 本发明化合物1-5的hmbc与
1
h-1
h cosy相关信号图。
具体实施方式
[0015]
下面的实施例将对本发明予以进一步的说明,但并不因此而限制本发明。
[0016]
实施例1(1)烈味脚骨脆茎叶14.0 kg用甲醇提取3次(用量3
ꢀ×ꢀ
140 l),减压回收提取液,得粗提物;(2)步骤(1)所得甲醇提取物,加水制成混悬液,分别用乙酸乙酯和石油醚萃取,得乙酸乙酯和石油醚萃取物;(3)步骤(2)石油醚和乙酸乙酯萃取物分别经硅胶柱色谱法分离,依次以石油醚: 丙酮100: 0, 100: 2, 100: 4, 100: 6, 100: 8, 100: 11, 100: 16, 100: 23, 100: 30洗脱;(3)上述步骤(2)中所得的石油醚: 丙酮100: 2~100: 30流份经中压液相色谱(mplc)分离,以甲醇/水6: 4~9: 1为流动相梯度洗脱;(4)上述步骤(3)中所得甲醇/水6: 4~9: 1流份经hplc-ri分离,以甲醇/水60: 40~90: 10为流动相洗脱得到新化合物1(收率0.003%),2(收率0.006%),3(收率0.002%),4(收率0.002%),5(收率0.006%)。
[0017]
根据化合物1-5的理化性质和波谱数据鉴定了其结构(化合物1-5的结构见图1;化合物1-5的波谱图见图2-图23)。
[0018]
化合物1的结构鉴定数据如下:无色油状; [α] +43.2 (c 0.1, ch
2
cl
2
); ecd (ch
3
cn) 199 (δε 17.1), 237 (δε
ꢀ-
2.5) nm; ir v
max 3435, 2961, 2926, 2878, 1728, 1466, 1373, 1195, 1155, 1109, 994, 967 cm
−
1
;hresims m/z 471.2720 [m + na]
+ (calcd for c
26
h
40
nao
6
, 471.2723); 13
c nmr (100 mhz, cdcl
3
)和
1
h nmr (400 mhz, cdcl
3
)数据,见表1-表2; 化合物的hmbc相关信号见图24。该化合物的绝对构型通过运用tddft(时间密度泛函理论)方法进行ecd(电子圆二色谱)计算确定,将实验测出的ecd谱图与计算得到的对映异构体的ecd图谱进行比较,确定了该化合物的绝对构型为2r, 5s, 6s, 8r, 9r, 10s, 18s, 19r。
[0019]
化合物2的结构鉴定数据如下:无色油状; [α] +37.6 (c 0.7, ch
2
cl
2
);ecd (ch
3
cn) 201 (δε16.0), 234 (δε-2.6) nm; ir v
max 3470, 2966, 2930, 1728, 1468, 1372, 1226, 1006, 947, 893, 736 cm-1
; esims m/z 499 [m + na]
+
; hresims m/z 499.2672 [m + na]
+ (calcd for c
27
h
40
nao
7
, 499.2672); 13
c nmr (100 mhz, cdcl
3
)和
1
h nmr (400 mhz, cdcl
3
)数据,见表1-表2; 化合物的hmbc相关信号见图24。该化合物的绝对构型通过ecd计算确定,该化合物的绝对构型为2r, 5s, 6s, 8r, 9r, 10s, 18s, 19s。
[0020]
化合物3的结构鉴定数据如下:无色油状; [α]
ꢀ−
34.5 (c 0.4, ch
2
cl
2
); ecd(ch
3
cn) 195 (δε
ꢀ-
9.5), 213 (δε
ꢀ-
3.8), 228 (δε
ꢀ-
4.7) nm; ir (film) v
max 3468, 2963, 2934, 1729, 1373, 1226, 1006, 955, 891, 736 cm
−
1
; esims m/z 513 [m + na]
+
; hresims m/z 513.2824 [m + na]
+ (calcd for c
28
h
42
nao
7
, 513.2828); 13
c nmr (100 mhz, cdcl
3
)和
1
h nmr (400 mhz, cdcl
3
)数据,见表1-表2; 化合物的hmbc相关信号见图24。该化合物的绝对构型通过ecd计算确定,该化合物的绝对构型为2s, 5s, 6s, 8r, 9r, 10s, 18s, 19s。
[0021]
化合物4的结构鉴定数据如下:无色油状; [α]
ꢀ-
34.4 (c 0.6,ch
2
cl
2
); ecd (ch
3
cn) 195 (δε-6.4), 212 (δε-2.7), 227 (δε-3.5) nm; ir v
max 3479, 2960, 2929, 2879, 1733, 1452, 1372, 1177, 702, 671 cm
−
1
; esims m/z 471 [m + na]
+
; hresims m/z 471.2357 [m + na]
+ (calcd for c
25
h
36
nao
7
, 471.2359); 13
c nmr (100 mhz, cdcl
3
)和
1
h nmr (400 mhz, cdcl
3
)数据,见表1-表2; 化合物的hmbc相关信号见图24。该化合物的绝对构型通过ecd计算确定,该化合物的绝对构型为2s, 5s, 6s, 8r, 9r, 10s, 18s, 19s。
[0022]
化合物5的结构鉴定数据如下:无色油状; [α] +2.5 (c 0.9, ch
2
cl
2
); ecd (ch
3
cn) 195 (δε18.7), 233 (δε-3.4) nm;ir (film) v
max 3467, 2966, 2927, 1729, 1468, 1371, 1222, 1003, 953, 890, 735 cm
−
1
; esims m/z 499 [m + na]
+
; hresims m/z 499.2671 [m + na]
+ (calcd for c
27
h
40
nao
7
, 499.2672); 13
c nmr (100 mhz, cdcl
3
)和
1
h nmr (400 mhz, cdcl
3
)数据,见表1-表2; 化合物的hmbc相关信号见图24。该化合物的绝对构型通过ecd计算确定,该化合物的绝对构型为2r, 5s, 6s, 8r, 9r, 10s, 18s, 19r。
[0023]
表1 化合物1-5的
13
c nmr数据
表2 化合物1-5的
1
h nmr数据
100: 11, 100: 16, 100: 23, 100: 30洗脱;(4)上述步骤(3)中所得的石油醚: 丙酮100: 2~100: 30流份经中压液相色谱(mplc)分离,以甲醇/水7: 3~9: 1为流动相梯度洗脱;(5)上述步骤(4)中所得甲醇/水(8: 2)流份经hplc-ri分离,以乙腈/水70: 30~90: 10为流动相洗脱得到新化合物1(收率0.002%),2(收率0.005%),3(收率0.003%),4(收率0.002%),5(收率0.006%)。
[0026]
化合物1-5的结构鉴定方法见实施例1。
[0027]
实施例4烈味脚骨脆中新化合物1-5的细胞毒活性测试。
[0028]
(1)实验原理抗肿瘤活性测试采用mtt法。四甲基偶氮唑盐 [3-(4,5-dimethyl-2-thiazoyl)-2, 5-diphenyl-tetrazolium bromide, mtt],为一种能接受氢离子的黄色染料,可结合到活细胞线粒体的呼吸链中,在琥珀酸脱氢酶和细胞色素的作用下mtt 的四氮唑环开裂,生成兰紫色的formazan 结晶。formazan 结晶生成量仅与活细胞数目成正比,死细胞中此酶消失,不能将mtt还原。生成的formazan 结晶可在二甲基亚砜(dmso)中溶解,利用酶标仪在490 nm 处测溶液的光密度od值,od值的大小与所生成的formazan 结晶的量成正比,从而反映出药物对细胞增殖的影响。
[0029]
(2)实验方法
①ꢀ
肿瘤细胞的培养以1640培养基(人肺癌细胞a549)或dmem高糖培养基(人肝癌细胞hepg2)作为基础配制成内含10%胎牛血清及1%双抗(青霉素:链霉素=1:1)的细胞培养液,细胞于37℃,5% co
2
培养箱培养,至细胞基本铺满培养瓶瓶底,传代或实验处理。
[0030]
②ꢀ
化合物的配制方法待测化合物用dmso溶解,配成母液,浓度为30 mm,储存于-20℃。临用时用培养液将其进行稀释,依次稀释为60 μm、30 μm、6 μm,或依据情况稀释为更低的浓度。
[0031]
③ꢀ
待测化合物的细胞毒活性测试取指数生长期的细胞,调细胞密度为1
×
10
4
个/ml,取100 μl接种于96孔板中,置于37℃,5%的培养箱中,培养24 h后,加不同浓度的待测化合物处理,48 h后,每孔加入20
ꢀꢀ
浓度为5 mg/ml 的mtt溶液,4 h后弃上层培养液,加入150 μl dmso,震荡7 min,用酶标仪测490 nm处各孔的吸光值,根据吸光度值计算各化合物对肿瘤细胞增殖的抑制率。
[0032]
④ꢀ
ic
50
的计算方法将各剂量和抑制率参数用非线性回归拟合计算化合物抑制肿瘤细胞增殖的ic
50
值。
[0033]
(3)实验结果:化合物1-5的细胞毒活性ic
50
见表3。
[0034]
表3 新化合物1-5细胞毒活性ic
50
值
a 依托泊苷为阳性对照药物。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips