
 商标分类
商标分类  商标转让
商标转让 
一种铂配合物蓝光材料及其制备和应用的制作方法
 2021-02-02 12:02:23|
2021-02-02 12:02:23| 353|
353| 起点商标网
起点商标网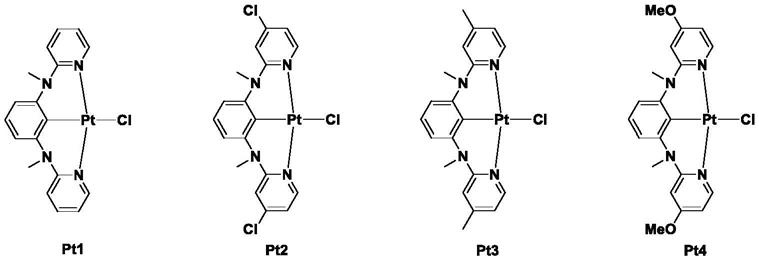
[0001]
本发明涉及光电材料技术领域,具体涉及的是一种铂配合物蓝光材料及其制备和在有机电致发光器件中的应用。
背景技术:
[0002]
在过去几十年里,发光金属配合物吸引了科学家广泛的研究兴趣。其中金属铂配合物作为一种重要的发光材料,被广泛应用于生物医药、有机电致发光器件、能量转换、化学传感器以及生物成像等领域。
[0003]
目前文献报道的发光金属铂配合物基本都具有平面四边形的空间构型,有利于发生分子间的pt
…
pt和π-π相互作用,因此其发光性质可以通过分子间相互作用调控。研究表明金属铂配合物中的分子间相互作用会使发光部分淬灭并发生红移现象,不利于开发发光性能优异的蓝光金属铂配合物及其发光器件。如何通过配体的设计,合成非平面的金属铂配合物,从而抑制分子间相互作用,成为提高金属铂配合物的蓝光发光性能的一种重要研究思路。
[0004]
从目前金属铂配合物发光材料的研究情况来看,性能优异的绿光以及红光金属铂配合物的研究已经较为成熟,其相应的发光器件的效率也较高。为了实现发光器件全色显示,蓝光材料、红光材料以及绿光材料都必不可少,但高效率、色度纯、结构简单的蓝光金属铂配合物还较少,因此发展性能优异的蓝光金属铂配合物具有很好的理论和应用价值。
技术实现要素:
[0005]
本发明的目的在于提供一种铂配合物蓝光材料,该材料具有优异的深蓝色发光性能。
[0006]
本发明的另一目的在于提供一种铂配合物蓝光材料的制备方法,制备方法简单,产率高。
[0007]
本发明的又一目的在于提供一种铂配合物蓝光材料在有机电致发光器件中的应用。
[0008]
为了达成上述目的,本发明的解决方案是:
[0009]
一种铂配合物蓝光材料,其结构通式如i所示,
[0010][0011]
其中,r
1
选自氢原子、氟原子、硝基、酯基以及三氟甲基中的一种,r
2
选自氢原子、氯原子、甲基以及甲氧基中的一种,r
3
选自甲基以及正丁基中的一种。
[0012]
下面列出优选的化合物(记为pt1~pt24)进一步阐述本发明一种铂配合物蓝光材
料,它们不应该被视为以任何方式限制本发明。
[0013]
[0014]
[0015][0016]
一种铂配合物蓝光材料的制备方法,包括以下步骤:
[0017]
(1)按摩尔比=1:2.1~3,称取间二溴苯衍生物化合物和n-烷基吡啶氨衍生物化合物
[0018]
(2)在氮气气氛下,向化合物a中加入添加剂、金属钯催化剂、碱,然后加入化合物b,再加入溶剂,将反应温度控制在100℃~150℃进行偶联反应,回流反应24~72h,所述添加剂和所述化合物a的摩尔比为0.01~0.1:1,所述金属钯催化剂和所述化合物a的摩尔比为0.01~0.1:1,所述碱和所述化合物b的摩尔比为3~6:1,所述溶剂的体积和所述化合物a的物质的量之比为5~20ml:1mmol;
[0019]
(3)待步骤(2)的反应结束后,冷却至室温,将产物依次进行抽滤、洗涤、过柱,得到中间产物化合物c
[0020]
(4)按摩尔比=1:1.5~2称取化合物c和化合物d k
2
ptcl
4
,在化合物c中加入化合物d,再加入溶剂,将反应温度控制在80℃~100℃进行金属配位反应,反应24~72h,所述溶剂的体积和所述化合物c的物质的量之比为6~10ml:0.1mmol;
[0021]
(5)待步骤(4)的反应结束后,冷却至室温,将产物依次进行抽滤,洗涤,烘干,得到所述铂配合物蓝光材料。
[0022]
步骤(2)中,所述金属钯催化剂为醋酸钯、二(三苯基膦)二氯化钯和三(二亚苄基丙酮)二钯中的一种;所述添加剂为1,1'-双(二苯基膦)二茂铁、三苯基磷或者三叔丁基磷;所述碱为叔丁醇锂或者叔丁醇钠。
[0023]
步骤(2)中,所述溶剂为1,4-二氧六环、甲苯和二甲苯中的一种。
[0024]
步骤(4)中,所述溶剂为冰醋酸或乙腈与水的混合溶剂中的一种。
[0025]
该铂配合物蓝光材料的合成路线如下所示:
[0026][0027]
一种铂配合物蓝光材料在有机电致发光器件中的应用,包括以下步骤:
[0028]
(1)制备如上所述的铂配合物蓝光材料;
[0029]
(2)在真空沉积系统中,依次将空穴传输层、发光层、电子传输层、电子注入层以及阴极材料蒸镀至ito基板上,干燥,氮气保护,得到有机电致发光器件;
[0030]
(3)采用keithley 2400参数分析仪、c9920-12外量子效率测量系统和光纤光谱仪pma-12分别对所述有机电致发光器件进行性能测试,且测得所述有机电致发光器件的最大外量子效率为1.8~3.7%;
[0031]
其中,所述发光层包括主体材料和所述铂配合物蓝光材料,所述主体材料选自mcp、dpepo和26mcpy中的一种,所述铂配合物蓝光材料的掺杂浓度为1wt%~10wt%,所述发光层的厚度为10~100nm。
[0032]
步骤(2)中,所述空穴传输层由mcp和moo
3
共混组成,其中moo
3
的掺杂浓度为10wt%~30wt%,mcp和moo
3
的浓度之和为100wt%,所述空穴传输层的厚度为20~50nm。
[0033]
步骤(2)中,所述电子传输层为tpbi,所述电子传输层的厚度为30nm,所述电子注入层为lif,所述电子注入层的厚度为0.7nm;所述阴极材料为al,所述阴极材料的厚度为100nm。
[0034]
步骤(2)中,所述ito基板在使用前,依次经过去污剂、去离子水以及乙醇超声清洗,然后经紫外臭氧清洗机清洗30~40min。
[0035]
采用上述技术方案后,本发明一种铂配合物蓝光材料是结构通式如i的化合物,单晶结构显示该化合物具有非平面的空间构型,分子间最短的pt
…
pt和芳香环之间的距离均为远远大于发生pt
…
pt和π-π相互作用所需要的空间距离,这表明分子间的pt
…
pt和π-π相互作用均得到了有效抑制,因此,本申请人通过在常用的三齿多吡啶配体[1,3-二(2-吡啶基)苯]的芳香环之间插入氨基,打破配体的共轭性,从而构建非平面的金属铂配合物,该材料具有优异的深蓝色发光性能。
[0036]
该铂配合物蓝光材料的制备方法简单,产率高达98%。采用真空蒸镀的方法,成功制备了该铂配合物蓝光材料掺杂的有机电致发光器件,且证明了该铂配合物蓝光材料在有机电致发光器件中具有良好的应用前景。
附图说明
[0037]
图1为为本发明化合物pt1的合成路线;
[0038]
图2为本发明化合物pt5的合成路线;
[0039]
图3为本发明化合物pt6的合成路线;
[0040]
图4为本发明化合物pt7的合成路线;
[0041]
图5为本发明化合物pt8的合成路线;
[0042]
图6为本发明化合物pt9的合成路线;
[0043]
图7为本发明化合物pt21的合成路线;
[0044]
图8为本发明化合物pt1的核磁氢谱图;
[0045]
图9为本发明化合物pt5的核磁氢谱图;
[0046]
图10为本发明化合物pt6的核磁氢谱图;
[0047]
图11为本发明化合物pt7的核磁氢谱图;
[0048]
图12为本发明化合物pt8的核磁氢谱图;
[0049]
图13为本发明化合物pt9的核磁氢谱图;
[0050]
图14为本发明化合物pt21的核磁氢谱图;
[0051]
图15为本发明化合物pt1、pt5-pt9和pt21在二氯甲烷稀溶液中的光谱图,其中(a)为吸收光谱,(b)为发射光谱图;
[0052]
图16为本发明化合物pt1、pt5-pt9和pt21在pmma薄膜中的发射光谱图;
[0053]
图17为本发明有机电致发光器件的性能测结果图,其中(a)为发射光谱,(b)为外量子效率图;
[0054]
图18为本发明化合物pt7在不同浓度条件下的二氯甲烷溶液中的发射光谱图。
具体实施方式
[0055]
为了进一步解释本发明的技术方案,下面通过具体实施例来对本发明进行详细阐述。
[0056]
一种铂配合物蓝光材料,其结构通式如i所示,
[0057][0058]
其中,r
1
选自氢原子、氟原子、硝基、酯基以及三氟甲基中的一种,r
2
选自氢原子、氯原子、甲基以及甲氧基中的一种,r
3
选自甲基以及正丁基中的一种。
[0059]
下面列出优选的化合物(记为pt1~pt24)进一步阐述本发明一种铂配合物蓝光材料,它们不应该被视为以任何方式限制本发明。
[0060]
[0061][0062]
一、铂配合物蓝光材料的制备
[0063]
实施例一
[0064]
化合物pt1的合成如图1所示:
[0065]
在氮气氛围下,将236mg(1.0mmol)1,3-二溴苯(化合物a)、92mg(0.1mmol)三(二亚苄基丙酮)二钯、56mg(0.1mmol)1,1'-双二苯基膦二茂铁以及634mg(6.6mmol)叔丁醇钠加入到耐压反应瓶中,抽放气两次之后,在氮气保护下将238mg(2.2mmol)2-(甲氨基)吡啶(化合物b)也加入至反应瓶中,再加入甲苯(20ml)并开始加热回流反应,温度控制在150℃,反应48h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、过柱,得到中间产物黄色油状液体(化合物c)220mg(产率76%),接着在氮气氛围下,称取87mg(0.3mmol)化合物c和187mg(0.45mmol)k
2
ptcl
4
(化合物d)至封管反应瓶中,加入乙腈和水的混合溶剂(v
乙腈
/v
水
=1:1)20ml并开始加热回流反应,温度控制在90℃,反应24h后停止反应,冷却至室温,将产物依次进行抽滤,洗涤,烘干,得到灰白色固体化合物pt1 99mg,产率:64%。
[0066]
实施例二
[0067]
化合物pt5的合成如图2所示:
[0068]
在氮气氛围下,将909mg(3.0mmol)1,3-二溴-5-三氟甲苯(化合物a)、276mg(0.3mmol)三(二亚苄基丙酮)二钯、168mg(0.3mmol)1,1'-双二苯基膦二茂铁以及1902mg(19.8mmol)叔丁醇钠加入到耐压反应瓶中,抽放气两次之后,在氮气保护下将713mg(6.6mmol)2-(甲氨基)吡啶(化合物b)也加入至反应瓶中,再加入甲苯(20ml)并开始加热回流反应,温度控制在160℃,反应72h|后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、过柱,得到中间产物黄色油状液体(化合物c)900mg(产率83%),在氮气氛围下,称取72mg(0.2mmol)化合物c和125mg(0.3mmol)k
2
ptcl
4
(化合物d)至封管反应瓶中,加入乙腈和水的混合溶剂(v
乙腈
/v
水
=1:1)20ml并开始加热回流反应,温度控制在90℃,反应72h后停止反应,冷却至室温,将产物依次进行抽滤,洗涤,烘干,得到灰白色固体化合物pt5 82mg,产率:70%。
[0069]
实施例三
[0070]
化合物pt6的合成如图3所示:
[0071]
在氮气氛围下,将304mg(1.0mmol)1,3-二溴-5-三氟甲苯(化合物a)、11mg(0.05mmol)醋酸钯、10mg(0.05mmol)三叔丁基磷以及叔丁醇锂528mg(6.6mmol)加入到耐压反应瓶中,抽放气两次之后,在氮气保护下将312mg(2.2mmol)2-(甲氨基)-4-氯吡啶(化合物b)也加入至反应瓶中,再加入甲苯(15ml)并开始加热回流反应,温度控制在130℃,反应30h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、过柱,得到中间产物黄色油状液体(化合物c)308mg(产率72%),在氮气氛围下,称取63mg(0.15mmol)化合物c和125mg(0.3mmol)k
2
ptcl
4
(化合物d)至封管反应瓶中,加入乙腈和水的混合溶剂(v
乙腈
/v
水
=1:1)10ml并开始加热回流反应,温度控制在90℃,反应24h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、烘干,得到灰白色固体化合物pt6 67mg,产率:74%。
[0072]
实施例四
[0073]
化合物pt7的合成如图4所示:
[0074]
在氮气氛围下,将304mg(1.0mmol)1,3-二溴-5-三氟甲苯(化合物a)、46mg(0.05mmol)三(二亚苄基丙酮)二钯、28mg(0.05mmol)1,1'-双二苯基膦二茂铁以及叔丁醇钠634mg(6.6mmol)加入到耐压反应瓶中,抽放气两次之后,在氮气保护下将256mg(2.1mmol)2-(甲氨基)-4-甲基吡啶(化合物b)也加入至反应瓶中,再加入二甲苯(15ml)并开始加热回流反应,温度控制在130℃,反应24h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、过柱,得到中间产物黄色油状液体(化合物c)340mg(产率88%),在氮气氛围下,称取57mg(0.15mmol)化合物c和125mg(0.3mmol)k
2
ptcl
4
(化合物d)至封管反应瓶中,加入乙腈和水的混合溶剂(v
乙腈
/v
水
=1:1)15ml并开始加热回流反应,温度控制在90℃,反应24h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、烘干,得到灰白色固体化合物pt7 85mg,产率:92%。
[0075]
实施例五
[0076]
化合物pt8的合成如图5所示:
[0077]
在氮气氛围下,将304mg(1.0mmol)1,3-二溴-5-三氟甲苯(化合物a)、46mg(0.05mmol)三(二亚苄基丙酮)二钯、28mg(0.05mmol)1,1'-双二苯基膦二茂铁以及634mg(6.6mmol)叔丁醇钠加入到耐压反应瓶中,抽放气两次之后,在氮气保护下将304mg(2.2mmol)2-(甲氨基)-4-甲氧基吡啶(化合物b)也加入至反应瓶中,再加入二甲苯(20ml)
并开始加热回流反应,温度控制在130℃,反应24h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、过柱,得到中间产物黄色油状液体(化合物c)300mg(产率71%),在氮气氛围下,称取84mg(0.2mmol)化合物c和125mg(0.3mmol)k
2
ptcl
4
(化合物d)至封管反应瓶中,加入乙腈和水的混合溶剂(v
乙腈
/v
水
=1:1)20ml并开始加热回流反应,温度控制在90℃,反应24h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、烘干,得到灰白色固体化合物pt8 120mg,产率:98%。
[0078]
实施例六
[0079]
化合物pt9的合成如图6所示:
[0080]
在氮气氛围下,将380mg(1.5mmol)1,3-二溴-5-氟苯(化合物a)、46mg(0.05mmol)三(二亚苄基丙酮)二钯、28mg(0.05mmol)1,1'-双二苯基膦二茂铁以及1296mg(13.5mmol)叔丁醇钠加入到耐压反应瓶中,抽放气两次之后,在氮气保护下将486mg(4.5mmol)2-(甲氨基)吡啶(化合物b)也加入至反应瓶中,再加入甲苯(20ml)并开始加热回流反应,温度控制在130℃,反应42h后停止反应,冷却至室温,将产物依次进行抽滤,洗涤,过柱,得到中间产物黄色油状液体(化合物c)200mg(产率43%),在氮气氛围下,称取31mg(0.1mmol)化合物c和84mg(0.2mmol)k
2
ptcl
4
(化合物d)至封管反应瓶中,加入乙腈和水的混合溶剂(v
乙腈
/v
水
=1:1)10ml并开始加热回流反应,温度控制在90℃,反应24h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、烘干,得到灰白色固体化合物pt9 30mg,产率:50%。
[0081]
实施例七
[0082]
化合物pt21的合成如图7所示:
[0083]
在氮气氛围下,将236mg(1.0mmol)1,3-二溴苯(化合物a)、92mg(0.1mmol)三(二亚苄基丙酮)二钯、56mg(0.1mmol)1,1'-双二苯基膦二茂铁以及1268mg(13.2mmol)叔丁醇钠加入到耐压反应瓶中,抽放气两次之后,在氮气保护下将330mg(2.2mmol)2-(正丁基氨基)吡啶(化合物b)也加入至反应瓶中,再加入二甲苯(15ml)并开始加热回流反应,温度控制在150℃,反应48h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、过柱,得到黄色油状液体(化合物c)100mg(产率27%),在氮气氛围下,称取75mg(0.2mmol)化合物c和125mg(0.3mmol)k
2
ptcl
4
(化合物d)至封管反应瓶中,加入乙腈和水的混合溶剂(v
乙腈
/v
水
=1:1)20ml并开始加热回流反应,温度控制在90℃,反应48h后停止反应,冷却至室温,将产物依次进行抽滤、洗涤、烘干,得到灰白色固体化合物pt21 65mg,产率:54%。
[0084]
二、结构分析
[0085]
1、实施例一制备得到的化合物pt1的核磁氢谱图如图8所示,
1
h nmr(400mhz,cd
2
cl
2
):δ3.52(s,6h),6.70-6.75(m,4h),7.01-7.08(m,4h),7.74(t,j=8.0hz,2h),9.19(d,j=6.0hz,1h)。
[0086]
化合物pt1的飞行时间质谱maldi-ms(m/z):484.1for[m-cl]
+
,计算值484.1。
[0087]
化合物pt1的元素分析:c,41.58;h,3.43;n,10.97;计算值:c,41.58;h,3.30;n,10.78。
[0088]
表明:核磁氢谱图、核磁碳谱图、飞行时间质谱以及元素分析结果均与化合物pt1的结构式吻合。
[0089]
2、实施例二制备得到的化合物pt5的核磁氢谱图如图9所示,1h nmr(400mhz,cd
2
cl
2
):δ3.57(s,6h),6.82(t,j=6.4hz,2h),6.96(s,2h),7.07(d,j=8.4hz,2h),7.81(t,
j=8.0hz,2h),9.21(d,j=6.0hz,2h)。
[0090]
化合物pt5的飞行时间质谱maldi-ms(m/z):552.1for[m-cl]
+
,计算值552.1。
[0091]
化合物pt5的元素分析:c,38.53;h,2.72;n,9.52;计算值:c,38.82;h,2.74;n,9.53。
[0092]
表明:核磁氢谱图、核磁碳谱图、飞行时间质谱以及元素分析结果均与化合物pt5的结构式吻合。
[0093]
3、实施例三制备得到的化合物pt6的核磁氢谱图如图10所示,
1
h nmr(400mhz,cd
2
cl
2
):δ3.55(s,6h),6.81(dd,j=4.4hz and 2.0hz,2h),6.97(s,2h),7.08(s,2h),9.11(d,j=8.0hz,2h)。
[0094]
化合物pt6的飞行时间质谱maldi-ms(m/z):620.8for[m-cl]
+
,计算值620.0。
[0095]
化合物pt6的元素分析:c,33.53;h,2.18;n,8.60;计算值(pt6
·
h
2
o):c,33.82;h,2.39;n,8.30。
[0096]
表明:核磁氢谱图、核磁碳谱图、飞行时间质谱以及元素分析结果均与化合物pt6的结构式吻合。
[0097]
4、实施例四制备得到的化合物pt7的核磁氢谱图如图11所示,1h nmr(400mhz,cd
2
cl
2
):δ2.36(s,6h),3.53(s,6h),6.64(d,j=8.0hz,2h),6.86(s,2h),6.92(s,2h),9.01(d,j=4.0hz,2h)。
[0098]
化合物pt7的飞行时间质谱maldi-ms(m/z):579.9for[m-cl]
+
,计算值580.1。
[0099]
化合物pt7的元素分析:c,38.87;h,3.15;n,8.39;计算值(pt7
·
1.5h
2
o):c,39.23;h,3.61;n,8.71。
[0100]
表明:核磁氢谱图、核磁碳谱图、飞行时间质谱以及元素分析结果均与化合物pt7的结构式吻合。
[0101]
5、实施例五制备得到的化合物pt8的核磁氢谱图如图12所示,
1
h nmr(400mhz,cd
2
cl
2
):δ3.51(s,6h),3.93(s,6h),6.42-6.46(m,4h),6.91(s,2h),8.99(d,j=8.0hz,2h)。
[0102]
化合物pt8的飞行时间质谱maldi-ms(m/z):611.8for[m-cl]
+
,计算值612.1。
[0103]
化合物pt8的元素分析:c,38.00;h,3.21;n,8.74;计算值(pt8
·
h
2
o):c,37.87;h,3.33;n,8.41。
[0104]
表明:核磁氢谱图、核磁碳谱图、飞行时间质谱以及元素分析结果均与化合物pt8的结构式吻合。
[0105]
6、实施例六制备得到的化合物pt9的核磁氢谱图如图13所示,1h nmr(400mhz,cd
3
cn):δ3.49(s,6h),6.61(t,j=12hz,2h),6.81(t,j=8.0hz,2h),7.17(d,j=8.0hz,2h),7.83(t,j=8.0hz,2h),9.14(d,j=4.0hz,2h)。
[0106]
化合物pt9的飞行时间质谱maldi-ms(m/z):501.9for[m-cl]
+
,计算值502.1。
[0107]
化合物pt9的元素分析:c,40.04;h,3.00;n,10.45;计算值:c,40.19;h,3.00;n,10.42。
[0108]
表明:核磁氢谱图、核磁碳谱图、飞行时间质谱以及元素分析结果均与化合物pt9的结构式吻合。
[0109]
7、实施例七制备得到的化合物pt21的核磁氢谱图如图14所示,
1
h nmr(400mhz,cd
2
cl
2
):δ0.93(t,j=7.6hz,6h),1.46-1.52(m,4h),1.75-1.78(m,4h),3.92-4.03(m,4h),
6.76(t,j=8.0hz,4h),7.04(t,j=8.0hz,1h),7.09(d,j=8.4hz,2h),7.73(t,j=7.6hz,2h),9.18(d,j=6.0hz,2h)。
[0110]
化合物pt21的飞行时间质谱maldi-ms(m/z):568.4for[m-cl]
+
,计算值568.2。
[0111]
化合物pt21的元素分析:c,47.32;h,4.74;n,9.12;计算值:c,47.72;h,4.84;n,9.28。
[0112]
表明:核磁氢谱图、核磁碳谱图、飞行时间质谱以及元素分析结果均与化合物pt21的结构式吻合。
[0113]
三、发光性能测试
[0114]
如图15所示,金属铂配合物pt1、pt5-pt9和pt21在二氯甲烷稀溶液中的最大发射波长均在420nm左右,表明上述铂配合物蓝光材料在溶液中均具有优异的深蓝色发光性能。
[0115]
采用溶液旋涂法将金属铂配合物pt7和pt8制备成薄膜。如图16所示,pt7 pmma薄膜和pt8 pmma薄膜的最大发射波长分别为452nm和445nm,表明上述铂配合物蓝光材料的薄膜也具有深蓝色发光性能,薄膜的最大绝对量子效率为12.1%。
[0116]
四、该铂配合物蓝光材料在有机电致发光器件中的应用
[0117]
1、一种铂配合物蓝光材料在有机电致发光器件中的应用,包括以下步骤:
[0118]
(1)制备如实施例四所述的铂配合物蓝光材料,并将ito基板依次经过去污剂、去离子水以及乙醇超声清洗,然后经紫外臭氧清洗机清洗半小时,待用;
[0119]
(2)在真空沉积系统中,将空穴传输层(mcp:20wt%moo
3
)30nm、发光层(mcp:7wt%pt7)20nm、电子传输层(tpbi)30nm、电子注入层(lif)0.7nm以及阴极材料(al)100nm蒸镀至ito基板上,干燥,氮气保护,得到有机电致发光器件,待测;
[0120]
(3)采用keithley 2400参数分析仪、c9920-12外量子效率测量系统和光纤光谱仪pma-12对有机电致发光器件(记为器件i)进行性能测试。
[0121]
2、一种铂配合物蓝光材料在有机电致发光器件中的应用,包括以下步骤:
[0122]
(1)制备如实施例四所述的铂配合物蓝光材料,并将ito基板依次经过去污剂、去离子水以及乙醇超声清洗,然后经紫外臭氧清洗机清洗半小时,待用;
[0123]
(2)在真空沉积系统中,将空穴传输层(mcp:20wt%moo
3
)30nm、发光层(mcp:1wt%pt7)20nm、电子传输层(tpbi)30nm、电子注入层(lif)0.7nm以及阴极材料(al)100nm蒸镀至ito基板上,干燥,氮气保护,得到有机电致发光器件,待测;
[0124]
(3)采用keithley 2400参数分析仪、c9920-12外量子效率测量系统和光纤光谱仪pma-12对有机电致发光器件(记为器件ii)进行性能测试。
[0125]
测试结果如图17所示,在器件i中,当铂配合物蓝光材料pt7的掺杂浓度为7wt%时,得到了天蓝色的发光器件,最大外量子效率为3.7%,在器件ii中,当铂配合物蓝光材料pt7的掺杂浓度为1wt%时,同样得到了天蓝色的发光器件,最大外量子效率为1.8%。且器件i和器件ii的光谱中在低能量区域(490~550nm)出现新的具有精细结构的发射峰,如图18所示,对处于二氯甲烷溶液中的铂配合物蓝光材料pt7的发射光谱进一步研究表明,当pt7的浓度超过1mm时,在490~554nm区域会产生新的具有精细结构的发射峰,与器件中的低能量区域的发射峰的位置基本一致,说明器件中的低能量发射峰有可能产生于分子间的相互作用。以上数据表明该化合物能够作为蓝色发光器件的候选材料,相信通过对主体材料以及器件结构的进一步优化,能够实现对性能优异的深蓝色发光器件的制备。
[0126]
上述实施例和图式并非限定本发明的产品形态和式样,任何所属技术领域的普通技术人员对其所做的适当变化或修饰,皆应视为不脱离本发明的专利范畴。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips