
 商标分类
商标分类  商标转让
商标转让 
2-羟基-5-壬基苯甲醛肟的新型合成方法与流程
 2021-02-02 10:02:44|
2021-02-02 10:02:44| 438|
438| 起点商标网
起点商标网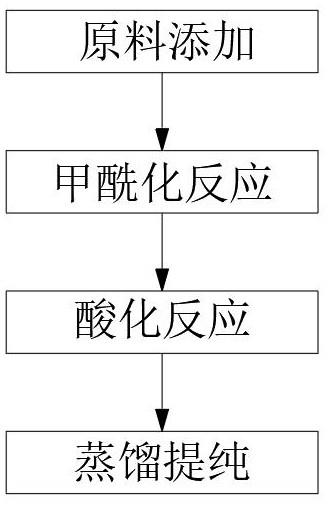
[0001]
本发明涉及化工萃取加工技术领域,具体为2-羟基-5-壬基苯甲醛肟的新型合成方法。
背景技术:
[0002]
多年来铜溶剂萃取主要基于两种主要的肟类萃取剂,醛肟与酮肟。两种肟类萃取剂因自身的优缺点比较鲜明,单独使用对铜的萃取一般效果不佳,通常会将2-羟基-5-壬基苯甲醛肟与2-羟基-5-壬基苯乙酮肟以一定比例进行复配或将2-羟基-5-壬基苯甲醛肟与酯改质剂进行复配后以获得最佳的萃取与反萃性能。
[0003]
由于长期铜源资的开采,高品位铜矿资源已越来越稀少,而对低品位铜矿资源的开发与利用推动着铜萃取技术发展,由于溶剂萃取技术日渐成熟,现国内外已有越来越多企业采用溶剂萃取方式对有价金属进行回收、提纯、分离。
[0004]
当前在全球范围内,羟肟类铜萃取剂在湿法冶炼铜行业已得到大规模应用。目前,世界上使用最多和最具代表性的铜萃取剂主要有德国巴斯夫产的lix系列、索尔维公司产的opt系列及m系列,这些系列的铜萃取剂主要活性成份中均采用了2-羟基-5-壬基苯甲醛肟,只是复配比例及方式不同而已。
[0005]
传统的加工方式会产生大量的气体造成环境的污染,同时合成的路径较长,工艺复杂,反应后的转化率和纯度较低,资源利用不足,造成原料的浪费和生产成本的提高。
[0006]
为此提供2-羟基-5-壬基苯甲醛肟的新型合成方法,以解决转化率、纯度和效率的问题。
技术实现要素:
[0007]
本发明的目的在于提供2-羟基-5-壬基苯甲醛肟的新型合成方法,以解决上述背景技术中提出的问题。
[0008]
为实现上述目的,本发明提供如下技术方案:2-羟基-5-壬基苯甲醛肟的新型合成方法,包括以下步骤:步骤一:原料添加,在反应瓶中分别加入壬基酚、甲苯、氯化镁、投料摩尔比为1:5.6:1.2,搅拌下滴加三乙胺76g并逐步升温;步骤二:甲酰化反应,将称量好多聚甲醛52.5g,分批次加入步骤一中的反应瓶中,每次3-4g,间隔时间为5-10分钟,多聚甲醛添加过程维持在3小时左右;步骤三:酸化反应,将步骤二中甲酰化反应后的反应产物降温度至60℃,然后将2.5n盐酸溶液缓慢加入步骤一中的反应瓶中,在温度为80℃的条件下保温反应1小时;步骤四:蒸馏提纯,将洗涤好有机相层先进行常压蒸馏,蒸出溶剂甲苯回收套用,再进行减压蒸馏。
[0009]
优选的,步骤一中所述壬基酚、甲苯、氯化镁的投料摩尔比为1:5.6:1.2,且在甲酰化反应时多聚甲醛与壬基酚的投料比为1:3.7。
[0010]
优选的,步骤一中所述三乙胺在搅拌5分钟后常温下滴加至反应瓶中,滴加完后开启加热装置逐步升温,瓶内液体颜色由白色变为深咖啡色,待瓶内产生回流,并且温度达到103-105℃时可进行甲酰化反应。
[0011]
优选的,步骤三所述的酸化反应保温1小时后的产物静置分相分出水相层,有机相层以水洗涤2遍,取有机相经核磁检测转化率不低于96.2%。
[0012]
优选的,步骤二中所述多聚甲醛添加完成后先加热蒸出瓶内部分甲醇,目的在于保持瓶内反应温度维持于103-105℃,在此条件下保温反应2小时。
[0013]
优选的,步骤四中的减压蒸馏中保持真空压力3-5mmhg,温度140℃-180℃条件下蒸出目标物2-羟基-5-壬基水杨醛。
[0014]
与现有技术相比,本发明的有益效果是:1、本发明的合成方法较以往甲醇-镁方法相比,合成路线更短、合成效率更高、无氢气产生,具有转化率高,纯度高等特点,可以与2-羟基-5-壬基苯乙酮肟及酯改质剂进行复配,以发挥其最佳的萃取与反萃性能;2、本发明采用三乙胺-氯化镁法为络合碱催化剂,一锅法合成得到2-羟基-5-壬基水杨醛,合成工艺路线短,可以获取很高的转化率和产品纯度。
附图说明
[0015]
图1为本发明合成方法流程图。
具体实施方式
[0016]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0017]
请参阅图1,实施例一,本发明提供一种技术方案,2-羟基-5-壬基苯甲醛肟的新型合成方法,该合成方法包括以下步骤:步骤一:原料添加,在反应瓶中分别加入壬基酚、甲苯、氯化镁、投料摩尔比为1:5.6:1.2,搅拌下滴加三乙胺76g并逐步升温;步骤二:甲酰化反应,将称量好多聚甲醛52.5g,分批次加入步骤一中的反应瓶中,每次3-4g,间隔时间为5-10分钟,多聚甲醛添加过程维持在3小时左右;步骤三:酸化反应,将步骤二中甲酰化反应后的反应产物降温度至60℃,然后将2.5n盐酸溶液缓慢加入步骤一中的反应瓶中,在温度为80℃的条件下保温反应1小时;步骤四:蒸馏提纯,将洗涤好有机相层先进行常压蒸馏,蒸出溶剂甲苯回收套用,再进行减压蒸馏。
[0018]
其中:壬基酚、甲苯、氯化镁的投料摩尔比为1:5.6:1.2,且在甲酰化反应时多聚甲醛与壬基酚的投料比为1:3.7;三乙胺在搅拌5分钟后常温下滴加至反应瓶中,滴加完后开启加热装置逐步升温,瓶内液体颜色由白色变为深咖啡色,待瓶内产生回流,并且温度达到103摄氏度时可进行甲酰化反应,酸化反应保温1小时后的产物静置分相分出水相层,有机
相层以水洗涤2遍,取有机相经核磁检测转化率不低于96.2%,多聚甲醛添加完成后先加热蒸出瓶内部分甲醇,目的在于保持瓶内反应温度维持于103摄氏度,在此条件下保温反应2小时,减压蒸馏中保持真空压力3mmhg,温度140℃条件下蒸出目标物2-羟基-5-壬基水杨醛。
[0019]
实施例二,本发明提供一种技术方案,2-羟基-5-壬基苯甲醛肟的新型合成方法,该合成方法包括以下步骤:步骤一:原料添加,在反应瓶中分别加入壬基酚、甲苯、氯化镁、投料摩尔比为1:5.6:1.2,搅拌下滴加三乙胺76g并逐步升温;步骤二:甲酰化反应,将称量好多聚甲醛52.5g,分批次加入步骤一中的反应瓶中,每次3-4g,间隔时间为5-10分钟,多聚甲醛添加过程维持在3小时左右;步骤三:酸化反应,将步骤二中甲酰化反应后的反应产物降温度至60℃,然后将2.5n盐酸溶液缓慢加入步骤一中的反应瓶中,在温度为100℃的条件下保温反应1小时;步骤四:蒸馏提纯,将洗涤好有机相层先进行常压蒸馏,蒸出溶剂甲苯回收套用,再进行减压蒸馏。
[0020]
其中:壬基酚、甲苯、氯化镁的投料摩尔比为1:5.6:1.2,且在甲酰化反应时多聚甲醛与壬基酚的投料比为1:3.7;三乙胺在搅拌5分钟后常温下滴加至反应瓶中,滴加完后开启加热装置逐步升温,瓶内液体颜色由白色变为深咖啡色,待瓶内产生回流,并且温度达到80摄氏度时可进行甲酰化反应,酸化反应保温1小时后的产物静置分相分出水相层,有机相层以水洗涤2遍,取有机相经核磁检测转化率不低于96.2%,多聚甲醛添加完成后先加热蒸出瓶内部分甲醇,目的在于保持瓶内反应温度维持于80℃,在此条件下保温反应2小时,减压蒸馏中保持真空压力4mmhg,温度150℃条件下蒸出目标物2-羟基-5-壬基水杨醛。
[0021]
实施例三,本发明提供一种技术方案,2-羟基-5-壬基苯甲醛肟的新型合成方法,该合成方法包括以下步骤:步骤一:原料添加,在反应瓶中分别加入壬基酚、甲苯、氯化镁、投料摩尔比为1:5.6:1.2,搅拌下滴加三乙胺76g并逐步升温;步骤二:甲酰化反应,将称量好多聚甲醛52.5g,分批次加入步骤一中的反应瓶中,每次3-4g,间隔时间为5-10分钟,多聚甲醛添加过程维持在3小时左右;步骤三:酸化反应,将步骤二中甲酰化反应后的反应产物降温度至60℃,然后将2.5n盐酸溶液缓慢加入步骤一中的反应瓶中,在温度为60℃的条件下保温反应1小时;步骤四:蒸馏提纯,将洗涤好有机相层先进行常压蒸馏,蒸出溶剂甲苯回收套用,再进行减压蒸馏。
[0022]
其中:壬基酚、甲苯、氯化镁的投料摩尔比为1:5.6:1.2,且在甲酰化反应时多聚甲醛与壬基酚的投料比为1:3.7;三乙胺在搅拌5分钟后常温下滴加至反应瓶中,滴加完后开启加热装置逐步升温,瓶内液体颜色由白色变为深咖啡色,待瓶内产生回流,并且温度达到120摄氏度时可进行甲酰化反应,酸化反应保温1小时后的产物静置分相分出水相层,有机相层以水洗涤2遍,取有机相经核磁检测转化率不低于96.2%,多聚甲醛添加完成后先加热蒸出瓶内部分甲醇,目的在于保持瓶内反应温度维持于120℃,在此条件下保温反应2小时,减压蒸馏中保持真空压力5mmhg,温度180℃条件下蒸出目标物2-羟基-5-壬基水杨醛。
[0023]
对比例:采用传统的甲醇-镁方法制备。
[0024]
实验后,通过对上述实施例一、实施例二和实施例三进行对比,得出实施例一条件下得到的产物最终检测的蒸馏收率为83.1%,核磁检测纯度为99.1%,产物的性能最佳,而实施例二和实施例三得到的产物蒸馏回收率低,纯度低于传统甲醇-镁方法得到的纯度,因此在实施例一的条件下采用三乙胺-氯化镁法为络合碱催化剂,一锅法合成得到2-羟基-5-壬基水杨醛,合成工艺路线短,可以获取很高的转化率和产品纯度,与对比例中以往甲醇-镁方法相比,合成路线更短、合成效率更高、无氢气产生,具有转化率高,纯度高的优点。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips