
 商标分类
商标分类  商标转让
商标转让 
一种双环醇二聚体、其制备方法及其用途与流程
 2021-02-02 10:02:13|
2021-02-02 10:02:13| 345|
345| 起点商标网
起点商标网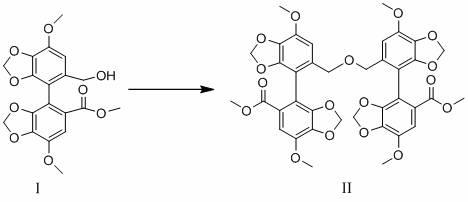
[0001]
本发明属于药物领域,具体涉及一种双环醇二聚体、其制备方法及其用途。
背景技术:
[0002]
肝病是一种影响我国人民生活质量的重大疾病,我国各种肝炎病毒携带者已超过一亿多人,研究开发临床疗效更好的肝病防治药物具有重要的意义。双环醇(bicyclol)是我国第一个拥有自主知识产权的一类抗肝炎创新药。卫生部在1996年12月批准双环醇进入临床试验,2001年09月双环醇获得了国家食品药品监督管理总局颁发的新药证书,获得了cfda颁发的双环醇的生产批文,由北京协和药厂生产上市。
[0003]
双环醇为白色或类白色结晶性粉末,无臭,无味,易溶于氯仿和丙酮,溶于乙腈,略溶于乙酸乙酯,微溶于乙醇,不溶于水。其商品名为百赛诺,其化学名为4,4'-二甲氧基-5,6,5',6'-双(亚甲二氧基)-2'-羟甲基联苯-2-甲酸甲酯,具有式i结构:在双环醇的制备过程中和在药品长期性的存放过程中会有杂质生成,杂质的存在不仅影响双环醇的纯度,还可能会带来非治疗性的毒副作用,因此对于双环醇杂质的结构或其合成方法的确定和利用合格的杂质标准品进行杂质对照对于有效控制双环醇原料药或其制剂的质量有重要意义。
技术实现要素:
[0004]
针对现有技术中存在的上述不足,尤其是因对双环醇杂质认识不足导致难以控制双环醇原料药或其制剂的质量的缺陷,本发明的目的在于提供一种双环醇二聚体、其制备方法及其用途,为双环醇原料药或其制剂的质量研究提供了有关物质对照品,对提高双环醇原料药或其制剂的质量标准具有重要的指导意义。
[0005]
为实现上述发明目的,本发明采用的技术方案如下:第一方面,本发明提供了一种双环醇二聚体,所述双环醇二聚体具有式ii结构:
上述双环醇二聚体的化学名称为二甲基5',5'''-(氧双(亚甲基))双(7,7'-二甲氧基-[4,4'-联苯并[d] [1,3]二氧杂环-5-羧酸酯),分子式为c
38
h
34
o
17
,分子量为762.67,该双环醇二聚体的确定对于有效控制双环醇原料药或其制剂的质量具有重要意义,可以应用于双环醇二聚体的定性、定量研究和检测,为双环醇原料药或其制剂的质量提供保证。
[0006]
第二方面,本发明提供了一种如第一方面所述的双环醇二聚体的制备方法,包括如下步骤:准备双环醇(式i)、反应溶剂和酸;双环醇(式i)在酸催化作用下于反应溶剂中加热进行反应,得到双环醇二聚体(式ii)。
[0007]
优选的是,在所述准备双环醇、反应溶剂和酸的步骤中,所述反应溶剂包括甲苯、四氢呋喃、乙腈、二氧六环中的至少一种,所述酸包括对甲苯磺酸、浓硫酸、甲磺酸中的至少一种,所述酸与所述双环醇的摩尔比为0.01~0.05,所述反应溶剂的体积与所述双环醇的质量比为5~15 ml/g。
[0008]
上述任一方案中优选的是,在所述准备双环醇、反应溶剂和酸的步骤中,所述反应溶剂的体积与所述双环醇的质量比为7~12 ml/g。
[0009]
上述任一方案中优选的是,在所述准备双环醇、反应溶剂和酸的步骤中,所述酸与所述双环醇的摩尔比为0.03~0.05。
[0010]
上述任一方案中优选的是,在所述双环醇在酸催化作用下于反应溶剂中加热进行反应,得到双环醇二聚体的步骤中,反应温度为70℃~110℃,反应时间为1h~4h。
[0011]
上述任一方案中优选的是,在所述双环醇在酸催化作用下于反应溶剂中加热进行反应,得到双环醇二聚体的步骤中,反应温度为80℃~100℃,反应时间为2h~3h。
[0012]
上述任一方案中优选的是,在所述双环醇在酸催化作用下于反应溶剂中加热进行
反应,得到双环醇二聚体的步骤后,还包括:对所述双环醇二聚体进行纯化。
[0013]
上述任一方案中优选的是,对所述双环醇二聚体进行纯化包括如下步骤:对含有所述双环醇二聚体的反应液减压浓缩至干,所得残留物经柱层析分离净化,收集洗脱液,对洗脱液进行减压浓缩,得到双环醇二聚体纯品。
[0014]
本发明提供的双环醇二聚体的制备方法中,以双环醇为原料,在酸催化作用下于反应溶剂中加热进行反应,经柱层析分离净化,得到双环醇二聚体纯品。该制备方法路线简单,操作方便,设备条件要求不高,极易实现,且后处理简单,同时该制备方法制得的双环醇二聚体纯度高,可以应用于双环醇二聚体的定性、定量研究和检测,对有效控制双环醇原料药或其制剂的质量有重要意义。
[0015]
第三方面,本发明提供了上述双环醇二聚体的用途,所述双环醇二聚体用于在双环醇有关物质检查时作为标准对照品。
附图说明
[0016]
图1为本发明实施例1制得的双环醇二聚体的核磁共振氢谱谱图;图2为本发明实施例1制得的双环醇二聚体的液相质谱谱图;图3为本发明实施例1制得的双环醇二聚体的高效液相色谱谱图;图4为本发明实施例1制得的双环醇二聚体的高效液相色谱谱图的峰表;图5为双环醇粗品与本发明实施例1制得的双环醇二聚体的高效液相色谱对照谱图;图6为图5中双环醇粗品的高效液相色谱图峰表;图7为双环醇另一粗品与本发明实施例1制得的双环醇二聚体的高效液相色谱对照谱图;图8为图7中双环醇粗品的高效液相色谱图峰表;图9双环醇又一粗品与本发明实施例1制得的双环醇二聚体的高效液相色谱对照谱图;图10为图9中双环醇粗品的高效液相色谱图峰表。
具体实施方式
[0017]
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0018]
除另有定义外,以下实施例中所用的技术术语具有与本发明所属领域技术人员普遍理解的相同含义。以下实施例中所用的实验试剂,如无特殊说明,均为常规生化试剂;所述实验试剂用量,如无特殊说明,均为常规实验操作中试剂用量;所述实验方法,如无特殊说明,均为常规方法。
[0019]
第一方面,本发明实施例提供了一种双环醇二聚体,所述双环醇二聚体具有式ii结构:
上述双环醇二聚体的化学名称为二甲基5',5'''-(氧双(亚甲基))双(7,7'-二甲氧基-[4,4'-联苯并[d] [1,3]二恶唑]-5-羧酸酯),分子式为c
38
h
34
o
17
,分子量为762.67,本申请发明人在对双环醇的制备工艺探究过程中,意外地发现双环醇粗品中有双环醇二聚体杂质,由高效液相色谱测定,双环醇二聚体的含量为0.25%左右,现有技术中没有对双环醇二聚体的相关报道,也无法从市场上购买得到双环醇二聚体标准品,该双环醇二聚体的确定对于有效控制双环醇原料药或其制剂的质量具有重要意义。药品中杂质含量的增加,会影响药品的纯度及其疗效且可能存在较大的安全隐患,将合格的双环醇二聚体标准品应用于双环醇二聚体的定性、定量研究和检测,可以为双环醇的安全用药提供保证。
[0020]
第二方面,本发明实施例还提供了一种如第一方面所述的双环醇二聚体的制备方法,包括如下步骤:(1)准备双环醇、反应溶剂和酸;(2)双环醇在酸催化作用下于反应溶剂中加热进行反应,得到双环醇二聚体。
[0021]
本发明实施例提供的双环醇二聚体的制备方法中,以双环醇为原料,在酸催化作用下于反应溶剂中加热进行反应,经柱层析分离净化,得到双环醇二聚体纯品。该制备方法路线简单,操作方便,设备条件要求不高,极易实现,且后处理简单,同时该制备方法制得的双环醇二聚体纯度高,可以应用于双环醇二聚体的定性、定量研究和检测,对有效控制双环醇及其制剂的质量有重要意义,为双环醇的安全用药提供了保证。
[0022]
进一步地,在所述准备双环醇、反应溶剂和酸的步骤中,所述反应溶剂包括甲苯、四氢呋喃、乙腈、二氧六环中的至少一种。其中,当选用四氢呋喃、乙腈和二氧六环做反应溶剂时,会产生较多的双环醇内酯,而得到的双环醇二聚体较少。但选用甲苯做反应溶剂时,会产生较少的双环醇内酯,较多的双环醇二聚体。双环醇内酯的合成路线如下图所示:
进一步地,在所述准备双环醇、反应溶剂和酸的步骤中,所述酸包括对甲苯磺酸、浓硫酸、甲磺酸中的至少一种。
[0023]
进一步地,在所述准备双环醇、反应溶剂和酸的步骤中,所述酸与所述双环醇的摩尔比为0.01~0.05,例如,酸与双环醇的摩尔比可以为0.01、0.02、0.03、0.04或0.05等。酸为催化剂,催化剂会对反应速度和反应深度产生影响,当酸的量过少时,会导致反应几乎不进行;当酸的量过多时,会导致副反应发生,生成的双环醇内酯增加,双环醇二聚体收率降低甚至无法制备得到双环醇二聚体。本发明实施例控制酸与双环醇的摩尔比为0.01~0.05,可以在较短的时间内,获得较多的双环醇二聚体,优选地,所述酸与所述双环醇的摩尔比为0.03~0.05,例如,酸与双环醇的摩尔比可以为0.03、0.04或0.05等,在此摩尔比下,得到的双环醇二聚体的收率较高。
[0024]
进一步地,在所述准备双环醇、反应溶剂和酸的步骤中,所述反应溶剂的体积与所述双环醇的质量比为5~15 ml/g,即每克双环醇需要的反应溶剂的体积为5ml~15ml,例如,反应溶剂的体积与双环醇的质量比可以为5ml/g、7ml/g、9ml/g、10ml/g、13ml/g或15ml/g等。反应溶剂会对反应速度和反应深度等产生影响,当反应溶剂的量过少时,原料浓度高,副反应多,产品收率较低;当反应溶剂的量过多时,会使反应原料的浓度降低,从而导致反应时间增加,产品的收率降低;本发明实施例中控制反应溶剂的体积与双环醇的质量的比为5~15 ml/g,可以在较短的反应时间内获得较多的双环醇二聚体,优选地,所述反应溶剂的体积与所述双环醇的质量比为7~12ml/g,例如反应溶剂的体积与双环醇的质量比可以为8ml/g、11ml/g或12ml/g等,在此比例下,得到的双环醇二聚体的收率较高。
[0025]
进一步地,在所述双环醇在酸催化作用下于反应溶剂中加热进行反应,得到双环醇二聚体的步骤中,反应温度为70℃~110℃,例如反应温度可以为70℃、80℃、90℃、100℃或110℃等。反应温度过低时,会导致反应时间变长,所述双环醇二聚体收率低;反应温度过高时,会导致副反应的发生,从而影响所述双环醇二聚体的收率,同时影响其纯度。而本实施例中控制反应温度在70℃~110℃之间,不仅可以保证反应时间较短,而且可以避免副反应的发生。优选地,所述反应温度为80℃~100℃,例如反应温度可以为85℃或95℃等,在此温度范围内,双环醇二聚体的收率较高。
[0026]
进一步地,在所述双环醇在酸催化作用下于反应溶剂中加热进行反应,得到双环醇二聚体的步骤中,反应时间为1h~4h,例如反应时间可以为1h、2h、3h或4h等。反应时间过短时,会出现反应后仍有原料剩余即反应不完全的现象,从而降低双环醇二聚体的收率,而本实施例中控制反应时间为1h~4h,可以保证在反应完全的同时双环醇二聚体的收率较高。优选地,所述反应时间为2h~3h,例如反应时间可以为2.2h、2.4h、2.6h或2.8h等。
[0027]
进一步地,在所述双环醇在酸催化作用下于反应溶剂中加热进行反应,得到双环醇二聚体的步骤后,还包括:对所述双环醇二聚体进行纯化。
[0028]
进一步地,所述对所述双环醇二聚体进行纯化包括如下步骤:对含有所述双环醇二聚体的反应液减压浓缩至干,所得残留物经柱层析分离净化,收集洗脱液,对洗脱液进行减压浓缩至干,得到双环醇二聚体纯品。所述柱层析分离净化过程中所用的硅胶柱目数为200~300目,洗脱剂为二氯甲烷和甲醇,二氯甲烷和甲醇的体积比为50:1。
[0029]
按照2015版《中国药典》方法中的的高效液相色谱法对其进行检测,按hplc(高效液相色谱)面积归一法计,确定双环醇二聚体的纯度,从检测结果可知得到的双环醇二聚体纯度为99.9%,纯度很高,因此利用该制备方法得到的双环醇二聚体可以作为标准品使用,可以应用于双环醇二聚体的定性、定量研究和检测,对有效控制双环醇及其制剂的质量有重要意义。
[0030]
本发明实施例的双环醇二聚体的制备方法的优选步骤为:(1)准备双环醇、甲苯和对甲苯磺酸,所述甲苯的体积与所述双环醇的质量的比为7~12 ml/g,所述对甲苯磺酸与所述双环醇与的摩尔比为0.03~0.05;(2)将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,控制反应温度为80℃~100℃,反应时间为2h~3h,得到双环醇二聚体,对含有所述双环醇二聚体的反应液减压浓缩至干,所得残留物经柱层析分离净化,收集含有产物的洗脱液,对洗脱液进行减压浓缩至干,得到双环醇二聚体纯品。
[0031]
第三方面,本发明实施例提供了上述双环醇二聚体的用途,所述双环醇二聚体用于在双环醇有关物质检查时作为标准对照品。
[0032]
本发明先后进行过多次试验,现举一部分试验结果作为参考,对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
[0033]
tlc(薄层色谱):下述实施例中tlc监控原料反应进度过程中,所用展开剂为二氯甲烷和甲醇,二氯甲烷和甲醇的体积比为10:1柱层析分离净化:本发明所述柱层析分离净化方法中均采用硅胶柱,硅胶柱目数为200~300目,洗脱剂为二氯甲烷和甲醇,二氯甲烷和甲醇的体积比为50:1。
[0034]
先按照本发明所述的双环醇二聚体的制备方法制备少许双环醇二聚体粗品,双环醇二聚体粗品经柱层析分离净化,收集不同时间点的洗脱液,经核磁共振氢谱谱图、质谱谱图和高效液相色谱谱图确认不同时间点流出的洗脱液中的物质结构,确认所述双环醇二聚体从硅胶柱中流出的时间点。
[0035]
实施例1步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸9.0mg(0.05mmol)。
[0036]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至100℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为3小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得白色固体390mg,其中,纯度为99.9%,收率为20%。
[0037]
对所得白色固体产物进行鉴定:白色固体产物的核磁共振氢谱谱图如图1所示,
1
hnmr(600mhz,dmso-d
6
)δ:7.22(s,1h),7.21(s,1h),6.60(s,1h),6.59(s,1h),6.05(s,2h),5.88-5.90(m,3h),5.84-5.86(m,3h),4.02-4.09(m,4h),3.91(s,3h),3.90(s,3h),3.82(s,3h),3.81(s,3h),3.51(s,3h),3.50(s,3h)。
[0038]
白色固体产物的质谱谱图如图2所示,检测仪器为waters 3100 mass detector,测试溶剂为甲醇,得到的白色固体产物的esi-ms(m/z):785.50 [m+na]
+
,本品分子量为
762.67,与本化合物分子量相符。
[0039]
因此可以确定上述白色固体产物为二甲基5',5'''-(氧双(亚甲基))双(7,7'-二甲氧基-[4,4'-联苯并[d] [1,3]二恶唑]-5-羧酸酯),其化学结构式如下所述:白色固体产物的纯度检测:取白色固体产物即双环醇二聚体0.5 mg溶解于1000 ml乙腈-水(乙腈和水的体积比为4:1)混合溶剂中,作为样品溶液,样品浓度为0.5 ug/ml,进样体积为5 ul,按照2015版《中国药典》方法中的高效液相色谱法对样品溶液进行测定,双环醇二聚体的高效液相色谱谱图及峰表如图3和图4所示,由检测结果可知白色固体产物的纯度为99.9%(2015版中国药典方法,按hplc面积归一法计)。
[0040]
实施例2步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、浓硫酸10mg(0.1mmol)。
[0041]
步骤(2),将准备好的甲苯、双环醇和浓硫酸依次加入到反应瓶中,搅拌并升温至80℃,双环醇在浓硫酸催化作用下于甲苯中进行反应,反应时间为2小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体290mg,其中,纯度为99.5%,收率为14.8%。
[0042]
实施例3步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸45mg(0.25mmol)。
[0043]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至70℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为1小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体420mg,其中,纯度为99.7%,收率为21%。
[0044]
实施例4步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸27mg(0.15mmol)。
[0045]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至100℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为3小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体460mg,其中,纯度为99.6%,收率为23%。
[0046]
实施例5步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸36mg(0.2mmol)。
[0047]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至100℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为3小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体480mg,其中,纯度为99.9%,收率为24%。
[0048]
实施例6步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸45mg(0.25mmol)。
[0049]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至100℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为3小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体500mg,其中,纯度为99.8%,收率为25%。
[0050]
实施例7步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸45mg(0.25mmol)。
[0051]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至80℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为3小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体440mg,其中,纯度为99.7%,收率为22%。
[0052]
实施例8步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸45mg(0.25mmol)。
[0053]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至90℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为3小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体460mg,其中,纯度为99.9%,收率为23%。
[0054]
实施例9步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸45mg(0.25mmol)。
[0055]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至90℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为2小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体440mg,其中,纯度为99.6%,收率为22%。
[0056]
实施例10步骤(1),20~30℃下,准备甲苯20ml、双环醇2.0g(5mmol)、对甲苯磺酸45mg(0.25mmol)。
[0057]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至90℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为4小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体500mg,其中,纯度为99.8%,收率为25%。
[0058]
实施例11步骤(1),20~30℃下,准备甲苯10ml、双环醇2.0g(5mmol)、对甲苯磺酸45mg(0.25mmol)。
[0059]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至90℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为3小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体340mg,其中,纯度为99.9%,收率为17%。
[0060]
实施例12步骤(1),20~30℃下,准备甲苯30ml、双环醇2.0g(5mmol)、对甲苯磺酸45mg(0.25mmol)。
[0061]
步骤(2),将准备好的甲苯、双环醇和对甲苯磺酸依次加入到反应瓶中,搅拌并升温至90℃,双环醇在对甲苯磺酸催化作用下于甲苯中进行反应,反应时间为3小时,tlc监控原料反应进度,反应完毕后,将反应液在40℃下减压浓缩至干,所得残留物经硅胶柱层析分离净化,收集含有产物的洗脱液,之后将洗脱液在45℃下减压浓缩至干,得双环醇二聚体白色固体440mg,其中,纯度为99.7%,收率为22%。
[0062]
试验例1验证双环醇粗品中是否存在双环醇二聚体取双环醇粗品,加乙腈-水(体积比为1:1)溶解并稀释制成每1ml中约含1mg的溶液,再精密量取1ml,置200ml量瓶中,用乙腈-水(体积比为1:1)稀释至刻度,摇匀,作为供试品溶液。称取双环醇二聚体标准品适量,作为对照品溶液。照高效液相色谱法(中国药典2020年版四部通则0512)试验,用十八烷基硅烷键合硅胶为填充剂(welch diamonsil plus c18 4.6mm
ꢀ×
250mm,5
µ
m或效能相当的色谱柱);以0.022%醋酸水溶液(量取纯化水900ml,加入200μl冰乙酸,充分混匀)为流动相a,以乙腈为流动相b,按下表程序进行梯度洗脱;流速为每分钟1.0ml,柱温40℃,检测波长为228nm。精密量取供试品溶液和对照品溶液5
µ
l,注入高效液相色谱仪,记录色谱图,如图5所示,双环醇粗品色谱图对应的峰表如图6所示。
[0063]
[0064]
图5为双环醇粗品和双环醇二聚体对照品的高效液相色谱图对照图,1号色谱图为双环醇粗品色谱图,2号色谱图为双环醇二聚体对照品色谱图,由图5可知,双环醇粗品中出现保留时间为12.652的双环醇二聚体的特征峰,与双环醇二聚体对照品的保留时间一致,因此双环醇粗品中存在双环醇二聚体杂质。
[0065]
由图6峰表可知,双环醇粗品中双环醇二聚体的含量为0.257%,远远高于药典2015版第二部中杂质含量不高于0.1%的规定。
[0066]
试验例2双环醇粗品中双环醇二聚体的定性分析取不同批次制备的双环醇粗品两份,按照试验例1的高效液相色谱检测方法,对所述两份双环醇粗品进行双环醇二聚体的定性分析,记录色谱图,如图7和9所示,图8为图7中双环醇粗品色谱图对应的峰表,图10为图9中双环醇粗品色谱图对应的峰表。1号色谱图为双环醇粗品色谱图,2号色谱图为双环醇二聚体对照品色谱图。
[0067]
由图7和图9可知,两份双环醇粗品中均存在与双环醇二聚体保留时间一致的色谱峰,说明两份双环醇粗品中均存在双环醇二聚体。
[0068]
由图8和图10峰表可知,双环醇粗品中双环醇二聚体的含量均为0.245%,远远高于药典2015版第二部中杂质含量不高于0.1%的规定。
[0069]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips