
 商标分类
商标分类  商标转让
商标转让 
2-乙基己酸铑(III)的制备的制作方法
 2021-02-02 09:02:04|
2021-02-02 09:02:04| 369|
369| 起点商标网
起点商标网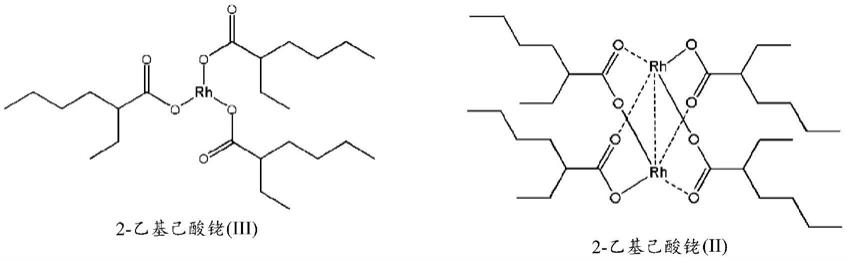
2-乙基己酸铑(iii)的制备
[0001]
本申请是原始中国专利申请号201680036038.1,申请日2016年7月1日,发明名称“2-乙基己酸铑(iii)的制备”的分案申请。
具体实施方式
[0002]
介绍
[0003]
本发明的主题是用于制备2-乙基己酸铑(iii)溶液的方法。2-乙基己酸铑(iii),rh[ch
3
(ch
2
)
3
ch(ch
2
ch
3
)coo]
3
,在下文中也被称为“rh(iii)2eh”。类似地,2-乙基己酸根被称为“2-eh”。
[0004]
根据本发明的方法的特征在于改善的过程执行。其有利于高收率和极高质量的rh(iii)2eh溶液的制备;此类溶液表现出极高的纯度。“高纯度”在此上下文中意味着低浓度的钠、氯、以及rh(ii)物质。相对于所使用的铑,收率超过99%。此外,根据本文提出的方法,在rh(iii)2eh的制备中空间收率比基于现有技术的已知方法的制备的空间收率高得多。此高空间收率意味着根据本发明的方法在工业规模上是经济上可行的。空间收率在此上下文中意味着在反应器中的每单位体积所形成的产物数量。
[0005]
根据本发明的rh(iii)2eh溶液特别适合在氢甲酰化反应中作为催化剂。
现有技术
[0006]
存在两种可能的2-乙基己酸铑结构:
[0007][0008]
2-乙基己酸铑的结构取决于铑的氧化程度。铑(ii)二聚物是绿色的,而铑(iii)化合物是黄棕色至红棕色。
[0009]
2-乙基己酸铑(iii)可以cas编号20845-92-5商购获得。
[0010]
铑羧酸盐主要是在化学工业中用作氢甲酰化反应的预催化剂。
[0011]
因此,wo 2009/059713a1公开一种通过利用一氧化碳的烯烃的氢甲酰化而制备醛类的方法,其中2-乙基己酸铑用作催化剂。氢甲酰化或羰氧化反应是过渡金属催化的利用氢和一氧化碳将烯烃或烯烃不饱和化合物转化成醛类和醇类,此类醛类和醇类所含有的碳原子比所用的烯烃多一个碳原子。同时,氢甲酰化过程获得了可观的商业及技术意义。例如,主要因此得到的醛类被原样使用或代表用于生产醇类、羧酸、酯类、或胺类的有价值的前体。
[0012]
氢甲酰化是由氢化金属羰基化物进行催化,有利地为元素周期表的viii族金属中
的那些。除了钴(经典的金属催化剂)以外,现在这几年基于铑的催化剂的使用越来越多。相对于钴,铑允许反应在较低的压力下进行。此外,当使用末端烯烃时,有利地形成直链正醛类,而异醛类仅占较小量。最终,基本上相较于钴催化剂的应用,在铑催化剂的存在下,使所用烯烃氢化成饱和烃显著降低。
[0013]
工业上,烯烃不饱和化合物的氢甲酰化是在以三级有机膦或亚磷酸酯作为配位体的铑羰基络合物的催化作用下进行。
[0014]
根据进一步的方法变化,铑催化的氢甲酰化反应也可以在不存在形成络合物的配位体(例如膦或亚磷酸酯)下进行。这种未使用膦或亚磷酸酯改性的铑催化剂及其作为氢甲酰化催化剂的适用性从文献中已知,且被称为未改性的铑催化剂。在专业文献中,假定铑化合物hrh(co)
4
在利用未改性的铑催化剂的氢甲酰化期间是催化活性的铑物质,虽然因为在反应区中许多化学反应彼此同时运行着,所以这个假定尚未被明确证实。据指出,在反应区中的氢甲酰化反应的条件下,未改性的铑催化剂由铑化合物形成,据称铑盐,诸如氯化铑(iii)、硝酸铑(iii)、乙酸铑(iii)、乙酸铑(ii)、硫酸铑(iii)、或铑(iii)氯化铵盐;来自铑含氧酸的盐,例如铑酸盐;来自铑羰基化合物,诸如rh
4
(co)
12
和rh
6
(co)
16
;或来自在一氧化碳/氢混合物(其也被称为合成气体)的存在下的有机铑化合物,诸如羰基乙酰丙酮铑、环辛二烯乙酸铑、或环辛二烯氯化铑。从而铑化合物可以固体使用或者适宜地在溶液中使用。例如,一种用于在未改性的铑络合物的存在下的氢甲酰化的方法,其中使用2-乙基己酸铑,描述于de 38 22 038a1中。
[0015]
用于铑溶液的制备的合适铑化合物是例如具有2至13个碳原子的脂族单羧酸盐或多羧酸盐。此外,铑的羰基化合物已被证明是非常成功的。虽然也可以使用卤素羰基化合物,但因为卤离子的腐蚀行为,其仅具有有限的应用。最终,铑的络合化合物,特别是铑(iii)的化合物,也是适合用于在催化剂系统中制备催化活性金属组分的起始材料。这些化合物含有单齿配位体、双齿配位体、或三齿配位体:诸如β-二酮,例如乙酰丙酮;或脂族不饱和烃和二乙烯属不饱和烃,诸如环戊二烯和1,5-环辛二烯。特别适合用于制备铑溶液的铑化合物是铑氧化物、铑羰基化物、乙酸铑、2-乙基己酸铑、异壬酸铑、以及乙酰丙酮铑(iii)。
[0016]
现有技术中已知两种用于制备2-乙基己酸铑的方法。
[0017]
us 4,845,306描述了一种方法,其中在第一容器中,将1.5当量naoh和1当量2-乙基己酸溶解于水中。在第二容器中,将氯化铑水合物溶解于水中。将1当量氯化铑水合物溶液添加到7当量2-乙基己酸钠溶液中,并在95℃下搅拌两小时。形成呈暗绿色油状物形式的粗产物。然后用酯醇-12提取该粗产物。在此方法中,铑的浓度在有机相中是10,000ppm,而在合并的水相中是2ppm,使得相对于所使用的铑,收率非常小。因为使用大量的过量碱,所以无法避免地形成了氢氧化铑。此外,绿色表示2-乙基己酸铑(ii)(rh(ii)2eh)的存在,而在催化中,2-乙基己酸铑(ii)被认为是活性较低的。
[0018]
wo 92/10460描述了一种方法,其中三氯化铑三水合物溶解于乙醇中。然后添加2-乙基己酸钠(na-2-eh)和乙基己酸,并且在室温下搅拌混合物。由此rhcl
3
*3h
2
o对na-2-eh的比率为大约1:3(mol/mol)。最后,将反应混合物加热至40℃,并过滤以分离所产生的nacl。得到黏稠的黄-绿色油状物。相对于所使用的铑,由此得到的收率是97%;然而,该油状物含有高浓度的钠离子和氯离子,并且绿色表示2-乙基己酸铑(ii)(rh(ii)2eh)的存在。由于在碱性介质中使用乙醇作为溶剂,可能会发生铑离子还原成铑金属的状况。出于此原因,需要
间歇过滤络合物以分离除去金属。
[0019]
在2-乙基己酸铑(iii)中的高含量的氯离子是不利的,因为氯离子是腐蚀性的,且通过降低催化剂的活性而在氢甲酰化中干扰催化反应。
[0020]
出于此原因,本发明的目的是克服现有技术在2-乙基己酸铑(iii)的制备中的缺点,并提供一种具有高收率的在工业规模上可执行的方法,并且该方法递送具有低钠离子和氯离子含量的反应产物。
[0021]
此问题是通过一种用于制备2-乙基己酸铑(iii)的溶液的方法解决,该方法包括以下步骤:
[0022]
a)通过在室温下在第一反应容器中将2-乙基己酸添加至碱金属氢氧化物水溶液而制备2-乙基己酸的金属盐的水溶液,其中2-乙基己酸对碱金属氢氧化物的摩尔比率为1.0:1.0至1.1:1.0(mol/mol),
[0023]
b)在第二反应容器中提供铑(iii)前体,该铑(iii)前体选自氯化铑(iii)水合物rhcl
3
*xh
2
o、氯化铑(iii)溶液h
3
[rhcl
6
]*(h
2
o)
n
、以及硝酸铑(iii)溶液rh(no
3
)
3
*(h
2
o)
n
、以及它们的混合物,
[0024]
c)将2-乙基己酸的碱金属盐的水溶液和铑(iii)前体的水溶液在反应容器中在20℃至30℃的内部温度下混合,
[0025]
d)加热来自步骤c)的混合物
[0026]-如果该rh(iii)前体为氯化铑(iii)溶液或氯化铑(iii)水合物,则在反应容器中加热到80℃至90℃的内部温度,或
[0027]-如果该rh(iii)前体为硝酸铑(iii),则加热到80℃至100℃的内部温度,
[0028]
e)在搅拌下冷却来自步骤d)的该悬浮液
[0029]-如果该rh(iii)前体为氯化铑(iii)溶液或氯化铑(iii)水合物,则冷却到40℃至50℃的内部温度,或
[0030]-如果该rh(iii)前体为硝酸铑(iii),则冷却到55℃至65℃的内部温度,
[0031]
f)在搅拌下添加不能与水混溶的醇、不能与水混溶的羧酸、或它们的混合物,
[0032]
g)后搅拌30分钟至3小时,
[0033]
h)冷却至室温,并保留乳液以使其沉降,
[0034]
i)排出下层不含rh的水相,
[0035]
j)如果在步骤中的该rh(iii)前体含有氯化铑(iii)水合物rhcl
3
*xh
2
o和/或氯化铑(iii)溶液h
3
[rhcl
6
]*(h
2
o)
n
,则用水性无机酸洗涤含有rh-2-eh的有机相。
[0036]
问题的解决方案包括提供用于制备2-乙基己酸铑(iii)的方法。由于所使用的化学品、过程方法、高产物质量、以及可实现的高收率、以及空间收率,该方法是环境友好且经济的。
[0037]
本方法包括2-乙基己酸铑(iii)的制备而无任何中间体的分离。本发明因此描述了其中从起始材料制备目标产物,而无昂贵且费时的中间体分离或中间体洗涤的过程。
[0038]
本方法提供了呈溶液形式的2-乙基己酸铑(iii)反应产物,其可直接用于rh(iii)2-eh应用作为催化剂的其它反应中。因此,不需要例如由于浓缩溶液或生产固体rh(iii)2-eh产生的昂贵且费时的分离。使用根据本发明的方法制备的rh(iii)2-eh溶液基本上不含2-乙基己酸铑(ii)。
[0039]
根据本发明的用于制备2-乙基己酸铑(iii)的方法说明如下,其中本发明包括下文单独列出的所有实施方案以及此类实施方案彼此的组合。
[0040]
在根据本发明的方法的步骤a)中,通过在搅拌下在室温下将2-乙基己酸添加至碱金属氢氧化物水溶液而制备2-乙基己酸的碱金属盐的水溶液。由此2-乙基己酸对碱金属氢氧化物的摩尔比率为1:1至1.1:1(mol/mol)。已发现1mol/l至6mol/l的碱金属氢氧化物水溶液的浓度是实际可用的。在一个具体的实施方案中,此碱金属氢氧化物水溶液是使用去矿物质水制备的。合适的碱金属氢氧化物为lioh、naoh、以及koh。有利地使用naoh。
[0041]
在根据本发明的方法的步骤b)中,提供铑(iii)前体。铑(iii)前体选自氯化铑(iii)水合物rhcl
3
*xh
2
o、氯化铑(iii)溶液h
3
[rhcl
6
]*(h
2
o)
n
、以及硝酸铑(iii)溶液rh(no
3
)
3
*2h
2
o、以及它们的混合物。任选地,在根据本发明的方法的步骤b)中,铑(iii)前体可用水稀释。
[0042]
对本领域技术人员而言,已知氯化铑(iii)水合物和氯化铑(iii)溶液并非是具有精确化学计量组成的限定化合物。因此,式rhcl
3
*xh
2
o、以及h
3
[rhcl
6
]*(h
2
o)
n
表示理想化的组成。本发明的络合化合物根据化合物的卤离子含量和水含量而变化。氯化铑(iii)水合物及其可商购获得的水溶液通常以混合的氯水(chloro-aquo)络合物存在,这是为什么在理想化式中的水含量被给定为“xh
2
o”的缘故。取决于氯化铑(iii)水合物及氯化铑(iii)溶液的生产过程,而会有更多或更少的水或氯离子配位体结合至铑(iii)络合物。在氯化铑(iii)水合物固体的生产中,这取决于蒸发的程度,而在氯化铑(iii)水合物溶液的生产中,这取决于酸含量(hcl)以及该溶液的浓度。
[0043]
待根据本发明使用的rh(iii)前体,氯化铑(iii)水合物rhcl
3
*xh
2
o,以及氯化铑(iii)溶液h
3
[rhcl
6
]*(h
2
o)
n
是可商购获得的。一般而言,所有的氯化铑(iii)水合物及氯化铑(iii)溶液可用于根据本发明的方法,无关于其各自的水或氯离子含量(rh/cl-比率),前提条件为这些氯化铑(iii)水合物及氯化铑(iii)完全溶于水。在本发明的上下文中,“完全溶于水”意味着在室温下,至少100g的对应铑化合物可溶于一升(1000ml)水中。
[0044]
在一个实施方案中,rh(iii)前体是氯化铑(iii)前体。其选自具有40%的最大铑含量的氯化铑(iii)水合物、并具有大约20%的铑含量和4:1至6:1的氯/铑比率的氯化铑(iii)溶液。
[0045]
如果氯化铑(iii)前体是h
3
[rhcl
6
]*n(h
2
o)(其在下文称为“氯化铑(iii)溶液”),则是特别有利的。一般而言,使用具有小于30重量%的铑含量的氯化铑(iii)水溶液,因为他们是可商购获得和生产可得的,例如通过在浓盐酸氯气的存在下使铑金属溶解。然而,合适的氯化铑(iii)溶液还可自贵金属回收或工业贵金属化学的过程流分流而得。此外,相较于通常使用的固体氯化铑(iii)水合物,使用氯化铑(iii)溶液具有提供更具有成本效益及更快处理的优点,因为上流蒸发、分离为氯化铑(iii)水合物以及分析以测定起始量不是必要的。
[0046]
在另外的实施方案中,氯化铑(iii)前体为氯化铑(iii)水合物rhcl
3
*xh
2
o。本领域技术人员已知此物质是固体。
[0047]
在另外的实施方案中,rh(iii)前体是具有15重量%的最大铑含量以及<0.1重量%的氯离子含量的硝酸铑(iii)溶液。这些类型的硝酸铑(iii)溶液是可商购获得的。另选地,硝酸铑溶液本身可以根据以下反应方程通过用硝酸将新鲜制备的氢氧化铑(iii)转
换成硝酸铑(iii)而制备:
[0048]
rhcl
3
+3naoh
→
rh(oh)
3
+3nacl
[0049]
rh(oh)
3
+3hno
3
→
rh(no
3
)
3
+3h
2
o
[0050]
获得rh(no
3
)
3
水溶液。此用于制备硝酸铑(iii)的方法对于本领域技术人员而言是已知的。
[0051]
待根据本发明使用的硝酸铑(iii)在上述定义的含义内也是完全水溶性的。
[0052]
如上所述,氯化铑(iii)溶液和硝酸铑(iii)以水溶液的形式存在。比之下,氯化铑(iii)水合物是固体。所有三种所提及的铑(iii)前体可用于根据本发明的方法的步骤b)中,无进一步地添加水或用水稀释、或在氯化铑(iii)水合物的情况下将该氯化铑(iii)水合物溶解于水中。去离子水,下文也称为“di水”有利地用于溶解或稀释铑(iii)前体。当然,去矿物质水或蒸馏水也是合适的。
[0053]
在一个具体实施方案中,在步骤b)中制备具有为15g/l至30g/l,有利地为20g/l至25g/l的纯铑浓度的rh(iii)前体水溶液。
[0054]
在根据本发明的方法的步骤c)中,在搅拌下,在20℃至30℃的反应容器的内部温度下,使来自步骤a)的2-乙基己酸的碱金属盐的溶液与来自步骤b)的rh(iii)前体水溶液混合。如此一来,2-乙基己酸(2-eh)的碱金属盐及rh(iii)前体相对于乙基己酸和纯铑的量以2:1至8:1(mol/mol)的比率有利地混合在一起。
[0055]
在一个具体实施方案中,rh(iii)前体为氯化铑(iii)溶液或氯化铑(iii)水合物,并且2-eh对rh(iii)前体的比率为6:1至8:1mol/mol。
[0056]
在一个另外的具体实施方案中,rh(iii)前体为硝酸铑(iii),并且2-eh对rh(iii)前体的比率为2:1至5:1mol/mol。
[0057]
2-eh溶液和rh(iii)前体溶液可连续混合或可不连续混合。连续混合意味着将2-eh溶液和rh(iii)前体溶液同时引入到混合容器中。不连续混合意味着将一种混合组分先完全引入,然后添加其它混合组分。
[0058]
在一个具体实施方案中,将rh(iii)前体溶液引入,然后添加碱金属2-eh溶液。
[0059]
在另一个有利的实施方案中,将碱金属2-eh溶液引入,然后提供rh(iii)前体溶液。
[0060]
一旦按照根据本发明的方法的步骤d)的2-eh溶液和rh(iii)前体溶液在搅拌下的混合结束,
[0061]-如果该rh(iii)前体为氯化铑(iii)溶液或氯化铑(iii)水合物,则将其在反应容器中加热到80℃至90℃的内部温度,或
[0062]-如果该rh(iii)前体为硝酸铑(iii),则将其加热到80℃至100℃的内部温度;
[0063]
并在此温度下搅拌1至4小时。有利地将其搅拌2至3小时。
[0064]
按照根据本发明的方法的步骤e),在搅拌下将悬浮液冷却,并确实执行以下步骤
[0065]-如果该rh(iii)前体为氯化铑(iii)溶液或氯化铑(iii)水合物,则冷却到40℃至50℃,更具体地45℃的内部温度,或
[0066]-如果该rh(iii)前体为硝酸铑(iii),则冷却到55℃至65℃,更具体地为60℃的内部温度。
[0067]
此后,按照根据本发明的方法的步骤f),在搅拌下添加不能与水混溶的醇或不能
与水混溶的羧酸、或它们的混合物。如果氯化铑(iii)水合物或氯化铑(iii)溶液被用作rh(iii)前体,则内部温度从而为40℃至50℃,而如果硝酸铑(iii)被用作rh(iii)前体,则内部温度从而为60℃至70℃。
[0068]
在本发明的范围内,如果在20℃,在水中醇类和羧酸的溶解度小于或等于50g/l,则此类醇类和羧酸被称为“不能与水混溶”。
[0069]
合适的醇类为具有5至12个碳原子的饱和脂族、芳族、以及芳脂族醇类,其在室温下是液体,诸如例如,戊-1-醇、戊-2-醇、戊-3-醇、2-甲基丁-1-醇、3-甲基丁-1-醇、2-甲基丁-2-醇、3-甲基丁-2-醇、2,2-二甲基丙-1-醇、己-1-醇、庚-1-醇、辛-1-醇、2,4,4-三甲基戊醇、壬-1-醇、3,3,5-三甲基己醇、3,5,5-三甲基己醇、癸-1-醇、十一-1-醇、十二-1-醇、戊-1,5-二醇、戊-1,5-二醇、1,2,3-丙三醇、环戊醇、苯基甲醇、1-苯基乙-1-醇、2-苯基乙-1-醇、酯醇-12、以及2,2,4-三甲基-1,3-戊二醇单异丁酸酯。酯醇-12为2,2,4-三甲基-1,3-戊二醇单异丁酸酯。也可使用这些醇类的混合物。
[0070]
合适的羧酸为具有5至13个碳原子的饱和羧酸,其在室温下是液体。正戊酸、2-甲基丁酸、正己酸、正庚酸、正辛酸、2-乙基己酸、正壬酸、异壬酸、以及异十三酸被提及作为示例。异壬酸和异十三酸的名称意指二异丁烯或四聚丙烯(tetrapropylene)经由氢甲酰化及随后的氧化而获得的反应产物。
[0071]
此处的“醇或羧酸或它们的混合物”意指
[0072]-单一醇或
[0073]-单一羧酸或
[0074]-数种醇类的混合物或
[0075]-数种羧酸的混合物或
[0076]-至少一种醇和至少一种羧酸的混合物
[0077]
可被使用。原则上,这意味着此种醇类和羧酸是按照上面的定义不能与水混溶。
[0078]
在一个具体实施方案中,醇或羧酸选自2-乙基己醇、2-乙基己酸、以及酯醇-12,其中在各情况下使用这些化合物中的单独一种。
[0079]
醇或羧酸、或它们的混合物用来:从步骤e)中所形成的悬浮液提取所形成的2-乙基己酸铑(iii)。在步骤f)中进行提取期间,2-乙基己酸铑(iii)基本上定量地转移到有机相中。醇或羧酸的量、或混合物的量,是在很广的范围内自由选择的。有利地选择,使得2-乙基己酸铑(iii)在此有机相中的浓度与在完全实现根据本发明的方法后应获得的2-乙基己酸铑(iii)的浓度大约相等或略高。根据本发明的方法提供了2-乙基己酸铑(iii)在醇、羧酸、或它们的混合物中的即用(ready-to-use)溶液,而这些即用溶液可以直接用作催化剂溶液,例如,如氢甲酰化反应中的催化剂溶液。如果按照步骤f)的在有机相中的铑2eh的浓度略高于在完全实现根据本发明的方法后应为的浓度,则该有机相中的铑2eh可从而在被用作催化剂溶液之前被稀释。有利地,在此处使用与在步骤f)中所使用的相同的醇或相同的羧酸、或相同的它们的混合物。
[0080]
如果按照根据本发明的方法的步骤d),所使用的rh(iii)前体是氯化铑(iii)溶液或氯化铑(iii)水合物,则温度一定不得超过90℃,且在步骤e)和f)中,温度一定不得超过50℃,原因是不依此原则就形成2-乙基己酸铑(iii)和2-乙基己酸铑(ii)的混合物。2-乙基己酸铑(ii)在下文中也称为rh(ii)2eh。在形成rh(ii)2eh的期间,形成绿色产物溶液。这显
示于比较例1至比较例3中。
[0081]
然而,如果所使用的rh(iii)前体为硝酸铑(iii),则在根据本发明的方法的步骤d)的温度可为高达100℃,并且在步骤e)和f)中,温度可为高达65℃,而无rh(ii)2-eh形成。
[0082]
来自步骤f)的反应混合物现在在相同的内部温度下,按照步骤g)再次搅拌30分钟至3小时,有利地再次搅拌1小时至2小时。
[0083]
按照根据本发明的方法的步骤h),然后将反应混合物冷却到20℃至30℃的内部温度。停止搅拌乳液,并使所形成的乳液沉降,其中发生了相分离。有利地,沉降在一小时至四小时的时间段内发生。
[0084]
然后将底部水相排出并弃除(步骤i)。
[0085]
如果氯化铑(iii)水合物rhcl
3
*xh
2
o和/或氯化铑(iii)溶液h
3
[rhcl
6
]*(h
2
o)
n
被用作rh(iii)前体,则随后用水性无机酸洗涤有机相至不含氯。合适的无机酸为不含卤素的酸,诸如,例如,硫酸、硝酸、以及磷酸。有利地,使用0.5%至2%无机酸水溶液,特别有利地,使用0.5%至2%硫酸水溶液。推荐的是,对于各洗涤阶段,使用与2-eh碱金属盐水溶液和rh(iii)前体水溶液的体积之和对应的大约一样多的无机酸溶液。在无机酸的每次添加后,在室温下进行搅拌2小时至6小时,随后使乳液保留以使该乳液沉降2小时至6小时,然后将底部水相排出并弃除。有利的是重复此洗涤步骤一次或两次。
[0086]
取决于所使用的铑(iii)前体,本方法提供基于金属计,以99%金属的收率(“金属收率”)的2-乙基己酸铑(iii)溶液。获得的rh(iii)2eh溶液基本上不含rh(ii)物质。这可在根据本发明的溶液的颜色中观察到:2-乙基己酸铑(iii)溶液具有黄棕色,而2-乙基己酸铑(ii)溶液是绿色的。在现有技术中引用的一些用于制备2-乙基己酸铑(iii)的方法提供含有显著比例的2-乙基己酸铑(ii)的产物。然而,rh(ii)2-eh在氢甲酰化反应中的催化效果比rh(iii)2eh差。出于此原因,在氢甲酰化反应中,有利的是使用几乎完全由rh(iii)2-eh组成的2-乙基己酸铑,并且如果可能的话,不含rh(ii)2eh。
[0087]
uv/vis光谱法可用于研究通过根据本发明的方法获得的溶液实际上是否基本上不含rh(ii)2eh。如果硝酸铑(iii)溶液被用作rh(iii)前体,则测量执行步骤i)后获得的溶液,或者如果氯化铑(iii)溶液或氯化铑(iii)水合物被用作rh(iii)前体,则测量执行步骤j)后获得的溶液。在这两种情况下,对于通过uv/vis光谱法的测量,测量具有1.9重量%至2.1重量%的铑浓度的溶液。本领域技术人员已知的是,铑含量可通过ms-icp测定。如果铑含量高于1.9重量%至2.1重量%,则预先相应地调整溶液。合适的用于调整的溶剂为上述羧酸和醇类。有利地,使用与在根据本发明的方法的步骤f)中所使用的相同的羧酸、相同的醇、或相同的混合物来调整。
[0088]
通过uv/vis光谱法,在2mm qs比色管中,在597nm测量具有1.9重量%至2.1重量%铑含量的溶液。如果吸收谱带的强度小于或等于0.350,则铑(iii)2-eh溶液在本发明的含义范围内“基本上不含rh(ii)2eh”。
[0089]
如果氯化铑(iii)水合物和/或氯化铑(iii)溶液被用作rh(iii)前体,则根据本发明的rh(iii)2-eh溶液的钠含量低于500ppm,且总氯含量低于2,500ppm。如果硝酸铑(iii)被用作rh(iii)前体,则按照根据本发明的方法获得的rh(iii)2eh溶液的钠含量低于250ppm,且氯离子含量低于250ppm。从而指定的ppm值是指铑含量。
[0090]
2-乙基己酸铑(iii)适合在氢甲酰化反应中用作催化剂。此类方法在现有技术中
是已知的,且一般可被描述为包括以下步骤的用于氢甲酰化反应的方法:
[0091]-根据权利要求1至5中任一项所述提供2-乙基己酸铑(iii)或其溶液;
[0092]-在氢甲酰化反应中采用如此获得的2-乙基己酸铑(iii)或其溶液作为催化剂。
[0093]
实施例
[0094]
下文中,“去离子水”被称为“di水”。
[0095]
实施例1:在85℃下在2-乙基己醇中由氯化铑溶液制备2-乙基己酸铑(iii)
[0096]
在搅拌下,将19.3g的氢氧化钠(6.4eq.,99%,merck)溶解于100ml的di水中。在冷却至室温后,将71g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0097]
同时,在搅拌下,在1l双夹套反应器中,将呈大约39g的氯化铑(iii)溶液(umicore产品编号68.2565.2720;rh含量19.69重量%,cl/rh=4.86)形式的7.7g的rh稀释于350ml的di水中。
[0098]
在室温下,通过滴液漏斗于15分钟内将乙基己酸钠溶液添加到氯化铑溶液。然后将反应混合物加热至t
内部
:85℃并保持在此温度3小时。在3小时结束时,将反应混合物冷却至t
内部
:45℃。在此温度下,通过滴液漏斗添加312g的2-乙基己醇(98%,biesterfeld)。然后再次搅拌现在呈棕-黄色的乳液2小时,并且随后冷却至25℃。然后关闭搅拌器,并且保留乳液用于沉降一小时。发生相分离。将底部无色的水相排出。将400ml的0.7%h
2
so
4
水溶液添加到有机相,并使乳液搅拌4小时。4小时后,将搅拌器关闭,并且相分离在1小时内再次发生。然后将水相排出。再次重复洗涤步骤。
[0099]
以99%金属收率获得具有大约2%rh的澄清黄-棕色产物溶液。
[0100]
总氯含量通过氯分析器测定,并且为1350ppm(相对于铑)。钠含量根据icp-oes测定,钠含量为<500ppm(相对于铑)。uv/vis光谱显示在597nm处强度为0.242的吸收谱带。
[0101]
实施例2:在85℃下在2-乙基己酸中由氯化铑溶液制备2-乙基己酸铑(iii)
[0102]
在搅拌下,将19.3g的氢氧化钠(99%,merck)溶解于100ml的di水中。在冷却至室温后,将71g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0103]
同时,在搅拌下,在1l双夹套反应器中,将呈大约42g的氯化铑(iii)溶液(umicore产品编号68.2565.2720;rh含量18.39重量%,cl/rh=4.93)形式的7.7g的rh稀释于350ml的di水中。
[0104]
在室温下,通过滴液漏斗于15分钟内将乙基己酸钠溶液添加到氯化铑溶液。将反应混合物加热至t
内部
:85℃并保持在此温度3小时。在3小时结束时,将反应混合物冷却至t
内部
:45℃。在此温度下,通过滴液漏斗添加318g的2-乙基己酸(98%,oxea)。然后再次搅拌现在呈棕-黄色的乳液2小时,并且随后冷却至25℃。然后关闭搅拌器,并且保留乳液用于沉降一小时。发生相分离。将底部水相排出。将400ml的0.7%h
2
so
4
水溶液添加到有机相,并使乳液搅拌4小时。4小时后,将搅拌器关闭,并且相分离在1小时内再次发生。然后将水相排出。再次重复洗涤步骤。
[0105]
以99%收率获得具有大约2%rh的澄清黄-棕色产物溶液。
[0106]
总氯含量通过氯分析器测定,并且为2100ppm(相对于铑)。钠含量根据icp-oes测定;钠含量为<500ppm(相对于铑)。uv/vis光谱显示在597nm处强度为0.258的吸收谱带。
[0107]
实施例3:在85℃下在酯醇-12中由氯化铑溶液制备2-乙基己酸铑(iii)
[0108]
在搅拌下,将19.3g的氢氧化钠(99%,merck)溶解于100ml的di水中。在冷却至室
温后,将71g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0109]
同时,在搅拌下,在1l双夹套反应器中,将呈43g的氯化铑(iii)溶液(umicore产品编号68.2565.2720;rh含量17.83重量%,cl/rh=4.99)形式的7.7g的rh稀释于350ml的di水中。
[0110]
在室温下,通过滴液漏斗于15分钟内将乙基己酸钠溶液添加到氯化铑溶液。将反应混合物加热至t
内部
:85℃并保持在此温度3小时。在3小时结束时,将反应混合物冷却至t
内部
:45℃。在此温度下,通过滴液漏斗添加357g的酯醇-12(99%,2,2,4-三甲基-1,3-戊二醇单异丁酸酯,sigma aldrich)。然后再次搅拌现在呈棕-黄色的乳液2小时,并且随后冷却至25℃。然后关闭搅拌器,并且保留乳液用于沉降一小时。发生相分离。将底部水相排出。将400ml的0.7%h
2
so
4
水溶液添加到有机相,并使乳液搅拌4小时。4小时后,将搅拌器关闭,并且相分离在1小时内再次发生。然后将水相排出。再次重复洗涤步骤。
[0111]
以99%收率获得具有大约2%rh的澄清黄-棕色产物溶液。
[0112]
总氯含量通过氯分析器测定,并且为2300ppm(相对于铑)。钠含量根据icp-oes测定;钠含量为<500ppm(相对于铑)。uv/vis光谱显示在597nm处强度为0.324的吸收谱带。
[0113]
实施例4:在85℃下在2-乙基己醇中由硝酸铑溶液制备2-乙基己酸铑(iii)
[0114]
在搅拌下,将11g的氢氧化钠(99%,merck)溶解于150ml的di水中。在冷却至室温后,将40g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0115]
同时,在搅拌下,在1l双夹套反应器中,将呈大约90g的硝酸铑(iii)溶液(umicore产品编号68.2565.2810;rh含量10重量%,游离hno
3
/rh比率<2、cl含量<0.1%)形式的9.0g的rh稀释于150ml的di水中。
[0116]
在80℃下,通过滴液漏斗于15分钟内将乙基己酸钠溶液添加到硝酸铑溶液。然后将反应混合物加热至t
内部
:85℃并保持在此温度2小时。在2小时结束时,将反应混合物冷却至t
内部
:60℃。在此温度下,通过滴液漏斗添加400g的2-乙基己醇(98%,biesterfeld)。然后再次搅拌现在呈棕-黄色的乳液1小时,并且随后冷却至25℃。然后关闭搅拌器,并且保留乳液用于沉降三小时。发生相分离。将底部无色的水相排出。
[0117]
以99%金属收率获得具有大约2%rh的澄清黄-棕色产物溶液。
[0118]
总氯含量通过氯分析器测定,并且为1290ppm(相对于铑)。钠含量根据icp-oes测定;钠含量为<500ppm(相对于铑)。uv/vis光谱显示在597nm处强度为0.085的吸收谱带。
[0119]
实施例5:在95℃下在2-乙基己醇中由硝酸铑溶液制备2-乙基己酸铑(iii)
[0120]
在搅拌下,将11g的氢氧化钠(99%,merck)溶解于150ml的di水中。在冷却至室温后,将40g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0121]
同时,在搅拌下,在1l双夹套反应器中,将呈大约90g的硝酸铑(iii)溶液(umicore产品编号68.2565.2810;rh含量10重量%,游离hno
3
/rh比率<2、cl含量<0.1%)形式的9g的rh稀释于150ml的di水中。
[0122]
在80℃下,通过滴液漏斗于15分钟内将乙基己酸钠溶液添加到硝酸铑溶液。然后将反应混合物加热至t
内部
:95℃并保持在此温度2小时。在3小时结束时,将反应混合物冷却至t
内部
:60℃。在此温度下,通过滴液漏斗添加400g的2-乙基己醇(98%,biesterfeld)。然后再次搅拌现在呈棕-黄色的乳液1小时,并且随后冷却至25℃。然后关闭搅拌器,并且保留乳液用于沉降三小时。发生相分离。将底部水相排出。
[0123]
以99%金属收率获得具有大约2%rh的澄清黄-棕色产物溶液。
[0124]
总氯含量通过氯分析器测定,并且为1110ppm(相对于铑)。钠含量根据icp-oes测定;钠含量为<500ppm(相对于铑)。uv/vis光谱显示在597nm处强度为0.093的吸收谱带。
[0125]
比较例1:在95℃下在2-乙基己醇中由氯化铑溶液制备2-乙基己酸铑(iii)
[0126]
在搅拌下,将19.3g的氢氧化钠(99%,merck)溶解于100ml的di水中。在冷却至室温后,将71g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0127]
同时,在搅拌下,在1l双夹套反应器中,将呈大约39.6g的氯化铑(iii)溶液(umicore产品编号68.2565.2720;rh含量19.40重量%,cl/rh=4.74)形式的7.7g的rh稀释于350ml的di水中。
[0128]
在室温下,通过滴液漏斗于15分钟内将乙基己酸钠溶液添加到氯化铑溶液。将反应混合物加热至t
内部
:95℃并保持在此温度3小时。在3小时结束时,将反应混合物冷却至t
内部
:45℃。在此温度下,通过滴液漏斗添加312g的2-乙基己醇(98%,biesterfeld)。然后再次搅拌现在呈绿-黄色的乳液2小时,并且随后冷却至25℃。然后关闭搅拌器,并且保留乳液用于沉降一小时。发生相分离。将底部水相排出。将400ml的0.7%h
2
so
4
水溶液添加到有机相,并使乳液搅拌4小时。4小时后,将搅拌器关闭,并且相分离在1小时内再次发生。然后将水相排出。再次重复洗涤步骤。
[0129]
以99%金属收率获得具有大约2%rh的澄清暗绿色产物溶液。
[0130]
总氯含量通过氯分析器测定,并且为1090ppm(相对于铑)。钠含量根据icp-oes测定;钠含量为<500ppm(相对于铑)。uv/vis光谱显示在597nm处强度为0.465的吸收谱带。
[0131]
比较例2:在95℃下在2-乙基己酸中由氯化铑溶液制备2-乙基己酸铑(iii)
[0132]
在搅拌下,将19.3g的氢氧化钠(99%,merck)溶解于100ml的di水中。在冷却至室温后,将71g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0133]
同时,在搅拌下,在1l双夹套反应器中,将呈40g的氯化铑(iii)溶液(umicore产品编号68.2565.2720;rh含量19.28重量%,cl/rh=4.88)形式的7.7g的rh稀释于350ml的di水中。
[0134]
在室温下,通过滴液漏斗于15分钟内将乙基己酸钠溶液添加到氯化铑溶液。将反应混合物加热至t
内部
:95℃并保持在此温度3小时。在3小时结束时,将反应混合物冷却至t
内部
:45℃。在此温度下,通过滴液漏斗添加318g的2-乙基己酸(98%,oxea)。然后再次搅拌现在呈绿色的乳液2小时,并且随后冷却至25℃。然后关闭搅拌器,并且保留乳液用于沉降一小时。发生相分离。将底部水相排出。将400ml的0.7%h
2
so
4
水溶液添加到有机相,并使乳液搅拌4小时。4小时后,将搅拌器关闭,并且相分离在1小时内再次发生。然后将水相排出。再次重复洗涤步骤。
[0135]
以99%收率获得具有大约2%rh的澄清暗绿色产物溶液。
[0136]
总氯含量通过氯分析器测定,并且为2450ppm(相对于铑)。钠含量根据icp-oes测定;钠含量为<500ppm(相对于铑)。uv/vis光谱显示在597nm处强度为0.526的吸收谱带。
[0137]
比较例3:在95℃下在酯醇-12中由氯化铑溶液制备2-乙基己酸铑(iii)
[0138]
在搅拌下,将19.3g的氢氧化钠(99%,merck)溶解于100ml的di水中。在冷却至室温后,将71g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0139]
同时,在搅拌下,在1l双夹套反应器中,将呈大约39g的氯化铑(iii)溶液(umicore
产品编号68.2565.2720;rh含量19.67重量%,cl/rh=4.99)形式的7.7g的rh稀释于350ml的di水中。
[0140]
在室温下,通过滴液漏斗于15分钟内将乙基己酸钠溶液添加到氯化铑溶液。将反应混合物加热至t
内部
:95℃并保持在此温度3小时。在3小时结束时,将反应混合物冷却至t
内部
:45℃。在此温度下,通过滴液漏斗添加357g的酯醇-12(99%,2,2,4-三甲基-1,3-戊二醇单异丁酸酯,sigma aldrich)。然后再次搅拌现在呈绿色的乳液2小时,并且随后冷却至25℃。然后关闭搅拌器,并且保留乳液用于沉降一小时。发生相分离。将底部水相排出。将400ml的0.7%h
2
so
4
水溶液添加到有机相,并使乳液搅拌4小时。4小时后,将搅拌器关闭,并且相分离在1小时内再次发生。然后将水相排出。再次重复洗涤步骤。
[0141]
以99%收率获得具有大约2%rh的澄清暗绿色产物溶液。
[0142]
总氯含量通过氯分析器测定,并且为1300ppm(相对于铑)。钠含量根据icp-oes测定;钠含量为<500ppm(相对于铑)。uv/vis光谱显示在597nm处强度为0.748的吸收谱带。
[0143]
比较例4:根据us 4,845,306 a1,在酯醇-12中由氯化铑水合物制备2-乙基己酸铑(iii)
[0144]
在搅拌下,在1l双夹套反应器中,将32g的氢氧化钠(10.3eq.,99%,merck)溶解于400ml的di水中。在冷却至室温后,将78.4g的2-乙基己酸(98%,oxea)缓慢地逐滴添加。
[0145]
同时,在搅拌下,将呈大约39g的氯化铑(iii)水合物(umicore产品编号68.2562.1138;rh含量39.5重量%)形式的8g的铑溶解于360ml的di水中,然后在室温下,通过滴液漏斗于15分钟内添加到乙基己酸钠溶液。将反应混合物加热至t
内部
:95℃。氢氧化铑的黄色沉积物沉淀。产物的转换未发生。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签: rh

 热门咨询
热门咨询




tips