
 商标分类
商标分类  商标转让
商标转让 
一种电荷翻转两亲嵌段共聚物、制备方法、前体聚合物、纳米胶束和应用与流程
 2021-02-02 04:02:26|
2021-02-02 04:02:26| 354|
354| 起点商标网
起点商标网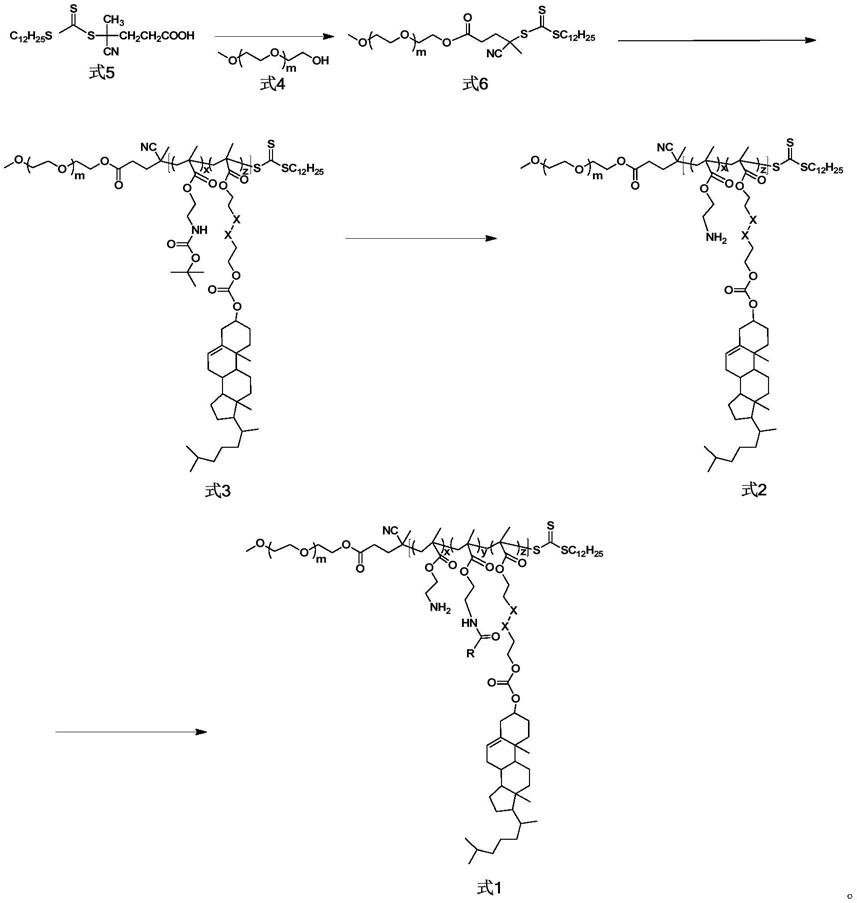
[0001]
本发明属于生物医用高分子材料技术领域,特别涉及具有电荷翻转特性的两亲嵌段共聚物以及基于该两亲嵌段共聚物的载药纳米胶束的制备及应用。
背景技术:
[0002]
癌症已成为21世纪人类健康的最大杀手,且其发病率和死亡率呈逐年上升的趋势,由此带来了沉重的经济和社会负担。近年来,科研工作者经探索制备得到了多种功能纳米材料,如纳米胶束、囊泡、微球、凝胶等,并应用作为小分子药物输送载体,以期解决现有药物欠佳的生物相容性、毒副作用、低靶向性和入胞困难等问题。相关研究虽已取得一定进展,但仍存在药物提前释放、体内稳定性差、疗效低、靶向功能不显著等缺陷,限制了其进一步临床应用。因此,如何解决目前药物载体普遍存在的体内循环时的隐蔽效应和对肿瘤组织的粘附效应间的矛盾,以及载体对药物的负载和特定部位靶向释放的矛盾,是目前高分子药物输送体系亟待解决的问题。
[0003]
近些年来,研究人员考虑引入酸敏基团对阳离子聚合物进行可逆修饰,使得载体在体内循环时带负电荷,有效避免与血液中蛋白的相互作用;到达肿瘤组织后,在弱酸环境中脱除修饰基团,使载体带正电荷,可增强载体与癌细胞的相互作用,提高载体入胞效率。此种具有电荷转换特性的聚合物可以同时赋予载体抗蛋白吸附性能并提高细胞摄取能力。另一方面,正常细胞外部谷胱甘肽(gsh)浓度较低(约2~20μm),而细胞内为2~10mm,因此,利用细胞内外gsh浓度的显著差异,在聚合物中引入具有还原响应性的二硫键,可实现负载药物的胞内响应性释放,提高生物利用率。
[0004]
然而,目前关于同时具有电荷翻转和还原响应性的聚合物报道较少,且已有的聚合物载体存在细胞相容性不佳、稳定性不足,或负载药物提前释放等问题。因此,采用天然生物相容性材料,构建具有良好体液稳定性、刺激响应性及电荷翻转行为的功能输送载体,实现药物的入胞效率和在特定部位的可控释放,是相关领域亟待解决的问题。
技术实现要素:
[0005]
本发明的目的在于提供一种具有ph响应电荷翻转特性的两亲嵌段共聚物及同时具有ph响应电荷翻转和还原响应的两亲嵌段共聚物药物载体,以及基于该载体的聚合物载药胶束。
[0006]
为实现上述目的,本发明提供了一种两嵌段共聚物,结构通式如式1所示:
[0007][0008]
其中r为基团或或x为c或者s,10≤m≤500;5≤x≤100;5≤y≤100;5≤z≤100,优选50≤m≤200;5≤x≤50;5≤y≤50;5≤z≤30
[0009]
为实现式1的制备,本发明提供了式1的前体聚合物,结构式如式2所示:
[0010][0011]
其中x为c或者s,10≤m≤500;5≤x≤200;5≤z≤100,优选50≤m≤200;5≤x≤100;5≤z≤30。
[0012]
为实现式2的制备,本发明提供了式2的前体聚合物,结构式如式3所示:
[0013][0014]
其中x为c或者s,10≤m≤500;5≤x≤200;5≤z≤100,优选50≤m≤200;5≤x≤100;5≤z≤30。式3是通过可逆加成-断裂链转移(raft)自由基聚合制备得到的。
[0015]
本发明的另一目的是提供式1、式2及式3所述嵌段共聚物的制备方法,其反应式如下图所示:
[0016][0017]
其中r为基团或或x为c或者s,10≤m≤500;5≤x≤100;5≤y≤100;5≤z≤100。
[0018]
具体制备方法为:
[0019]
步骤一:由端羟基聚乙二醇(式4)和raft链转移剂(式5)进行共价偶联,得到式6所示结构的化合物;
[0020]
步骤二:以式6所示结构的化合物作为大分子链转移剂,引发含有胆固醇砌块的单体(式7)和含有保护氨基的单体(式8)进行raft无规共聚,得到式3所示嵌段共聚物;
[0021]
步骤三:将式3所示嵌段共聚物脱除氨基的保护基后,得到式2所示结构的化合物;
[0022]
步骤四:通过式2所示结构的化合物末端氨基和酸酐进行反应,得到式1所示嵌段共聚物;
[0023]
其中10≤m≤500
[0024][0025]
其中10≤m≤500
[0026]
x为c或者s
[0027][0028]
本发明的另一目的是提供一种基于式1、式2及式3的胶束以及载药共聚物胶束的制备方法。
[0029]
具体为:将式1、式2或式3嵌段共聚物溶于有机溶剂a中,包括但不限于四氢呋喃、二甲基亚砜、二甲基甲酰胺、二甲基乙酰胺、二氧六环、丙酮以及两者的混合物,再将抗肿瘤药物溶解在有机溶剂b中,药物包括但不限于阿霉素、表阿霉素、紫杉醇、多西紫杉醇、喜树碱、5-氟尿嘧啶等,优选为阿霉素,有机溶剂b包括但不限于四氢呋喃、二甲基亚砜、二甲基甲酰胺、二甲基乙酰胺、二氧六环、丙酮、乙醇、甲醇、丁醇。随后,将嵌段共聚物溶液和药物溶液混合,再向混合溶液中逐滴滴加去离子水,待体系稳定后,将自组装溶液放入纤维素透析袋中(截留分子量3500da)透析48h,得到载药共聚物胶束溶液。
[0030]
本发明的另一目的是提供上述载药共聚物胶束的应用。该载药共聚物胶束具有响应性药物释放行为,包括ph响应性及还原响应性,能够有效进入细胞并抑制肿瘤细胞的增殖。
[0031]
有益效果
[0032]
本发明以聚乙二醇作为亲水嵌段,以含有胆固醇砌块单体和含有boc保护氨基单体的无规共聚物作为疏水嵌段,制备两亲嵌段共聚物。之后,脱除氨基保护基,并进一步后修饰引入β-羧基酰胺基团,制备得到具有ph响应电荷翻转特性的两亲嵌段共聚物。在一定条件下,该两亲嵌段共聚物自组装形成胶束,亲水性的聚乙二醇作为胶束的壳,含有胆固醇的疏水性嵌段作为胶束的核,包裹疏水性药物。在弱酸性环境下,β-羧基酰胺基团断裂,从而实现自组装胶束表面电荷由负向正的翻转,有效提高载体的入胞效率并实现药物的可控释放。同时,通过在含有胆固醇砌块的单体中引入二硫键,可以进一步实现药物在细胞内的还原响应性释放。
附图说明
[0033]
图1为本发明实施例1中的peg-b-p(machol-co-manboc)的核磁共振氢谱图;
[0034]
图2为本发明实施例1中peg-b-p(machol-co-manh2)的共聚物自组装胶束透射电镜图;
[0035]
图3为本发明实施例1中peg-b-p(machol-co-manh
2-dca)的载药共聚物胶束的粒径分布图;
[0036]
图4为本发明实施例1中共聚物自组装胶束的ph响应电荷翻转行为表征;
[0037]
图5为本发明实施例1中药物阿霉素的体外响应性释放行为;
[0038]
图6为本发明实施例1中载药共聚物胶束对mcf-7肿瘤细胞的增殖抑制效果图。
具体实施方式
[0039]
下面结合实施例和附图对本发明作进一步详细的说明。
[0040]
实施例1
[0041]
一种嵌段共聚物peg-b-p(machol-co-manh
2-dca)的制备方法,包括如下步骤:
[0042]
首先将raft链转移剂cdp和peg
5k
进行化学偶联,继而引发含有胆固醇砌块的单体machol和含有保护氨基的单体manboc无规共聚,然后在三氟乙酸中脱除氨基的保护基,生成末端氨基。进一步将该氨基在碱性条件下与四氢苯酐反应,制备得到侧链带有β-羧基酰胺基团的两亲嵌段共聚物,具体合成路线如下式所示:
[0043][0044]
步骤1:向干燥反应瓶中加入聚乙二醇-5k(2.5g,0.5mmol)、raft链转移剂cpadb
(1.67g,4mmol)、碳二亚胺(825mg,5mmol)和4-二甲氨基吡啶(122mg,1mmol),以及溶剂二氯甲烷(100ml),室温搅拌48h,过滤除去未溶解物质。真空浓缩后,在冷却的干乙醚中沉淀反应介质,并洗涤三次,真空干燥收集沉淀物,得到产物peg-cta。
[0045]
步骤2:将含胆固醇单体masschol(318mg,0.5mmol)、含保护氨基单体manboc(229mg,1mmol)和peg-cta(274.5mg,0.05mmol)置于氮气环境下的50ml schlenk管中。然后,将溶于无水甲苯(5ml)的偶氮二异丁氰(2.5mg,0.015mmol)注入反应瓶中。混合液经冷冻-解冻脱氧3次,然后浸入80℃恒温的油浴中反应12h后,液氮冷却停止反应,在冷却的干甲醇中沉淀并洗涤3次,收集沉淀物并真空干燥,得到peg-b-p(machol-co-manboc)。该共聚物的核磁氢谱表征如图1所示,从图中可以看出,peg-cta成功引发含有胆固醇的单体以及含有保护氨基的单体聚合,得到嵌段共聚物。
[0046]
步骤3:所制备的共聚物前体peg-b-p(machol-co-manboc)在三氟乙酸和二氯甲烷(1/2,v/v)混合溶液中室温脱保护8h,无水冷甲醇反复沉淀得到固体粉末,真空干燥。然后,将中间体重新溶于二氯甲烷溶液,再加入1,8-二氮杂双环[5.4.0]十一碳-7-烯(dbu,0.2当量)和3,4,5,6-四氢邻苯二甲酸酐(tda,5当量),室温反应24h。在冷却甲醇中沉淀3次,真空干燥过夜,得到产物peg-b-p(machol-co-manh
2-dca)。
[0047]
实施例2
[0048]
peg-b-p(machol-co-manh2)共聚物自组装胶束的制备,其步骤如下:
[0049]
将10.0mg嵌段共聚物溶于1ml thf中,并在室温下搅拌4h。然后,在搅拌下向混合物中逐滴加入5ml磷酸盐缓冲液(ph 7.4,10mm)。使用预膨胀纤维素透析膜(mwco 3500)将所得溶液在去离子水中进一步透析48h,得到peg-b-p(machol-co-manh2)共聚物自组装胶束。通过透射电镜对共聚物自组装胶束的粒径进行表征。
[0050]
从图2可以看出,共聚物在溶液中自组装得到粒径约为100nm左右的球,微球粒径分布较为均匀,表明成功通过自组装得到纳米胶束。
[0051]
实施例3
[0052]
阿霉素负载共聚物胶束的制备,其步骤如下:
[0053]
首先将10.0mg嵌段共聚物溶于1ml thf中,并在室温下搅拌4h。将阿霉素(10.0mg)和三乙胺(2mol当量)溶于1ml二甲基亚砜中,搅拌4h后,加入上述嵌段共聚物溶液中。然后在搅拌下向混合物中逐滴加入5ml磷酸盐缓冲液(ph 7.4,10mm)。使用预膨胀纤维素透析膜(mwco 3500)将所得溶液在去离子水中进一步透析48h,得到阿霉素负载共聚物胶束溶液。将获得的载药共聚物胶束在冻干机中冻干,通过动态光散射对胶束的粒径和粒径分布进行表征,通过紫外可见分光光度计测定阿霉素的负载量和包封率。
[0054]
从图3可以看出,载药共聚物胶束平均粒径为128nm,粒径分布0.168,表明成功制备得到分布较为均一的载药胶束。胶束对阿霉素的负载量约为12.8wt%,包封率约为82.1%。
[0055]
实施例4
[0056]
对上述共聚物自组装胶束ph响应性电荷翻转性能表征
[0057]
将共聚物自组装胶束分散到ph 6.0的缓冲溶液中,并在37℃下孵育,通过动态光散射实时监测胶束的粒径和zeta电位。
[0058]
如图4所示,在ph 6.0时,共聚物自组装胶束的zeta电位明显增加,1、4、8和24h后
分别达到-2.85、+2.45、+8.65和+10.98mv,表明β-羧基酰胺基团发生降解,由带负电荷的羧基转变为正电荷的氨基。同时,纳米粒子的尺寸在孵育4h后迅速增加到260.5nm,并在~150nm处稳定。尺寸的略微增加可能归因于β-羧基酰胺基团的解离导致的胶束亲水-疏水平衡的变化。纳米粒子的这种ph响应性电荷翻转特性有利于增强载药胶束和肿瘤细胞膜之间的相互作用,增强药物的内吞效率,进而提高疗效。
[0059]
实施例5
[0060]
对药物体外响应性释放行为进行实验
[0061]
5.0mg载药共聚物胶束分散于4ml缓冲溶液,装入透析袋内(截留分子量3500da),再放置于36ml对应缓冲溶液中,于37℃、150rpm摇床中孵育。四组缓冲溶液分别为ph 7.4,0mm谷胱甘肽(gsh),ph 6.0,0mm gsh,ph7.4,10mm gsh以及ph 6.5,10mm gsh的磷酸盐缓冲液。定时从透析袋外取2ml样品,并同步补充2ml新鲜缓冲溶液。测定取出溶液在λ=481nm处的紫外吸光度,并根据阿霉素在去离子水中浓度-吸光度标准工作曲线计算溶液中阿霉素浓度,由公式1进一步计算阿霉素累积释放量。
[0062][0063]
m
drug
表示胶束中药物质量,v0表示释放介质总体积(40ml),c
n
表示第n个样品中阿霉素浓度。
[0064]
由图5可见,阿霉素的释放行为呈现ph和还原双重响应特性。在ph 7.4、无gsh存在的条件下,药物释放速率很慢,48h药物累积释放量约为10%。在ph 6.0、无gsh存在的条件下,药物累积释放量有所增加,48h达到25%左右。当加入10mm gsh之后,在中性(ph 7.4)和弱酸性(ph 6.0)环境中药物阿霉素的释放速率和累积释放量都大大增强,且弱酸性条件下药物释放量更高,48h累积释放量达到70%。由此可见,该共聚物自组装胶束在体液环境中具有优良的稳定性,并且在肿瘤组织和细胞内环境中能够实现有效的响应性释放。
[0065]
实施例6细胞毒性实验
[0066]
首先将mcf-7细胞以6
×
103个细胞/孔的密度接种到96孔微孔板中,在含10%胎牛血清、丙酮酸钠(1mm)、青霉素(100iu/ml)和链霉素(100μg/ml)的100μl dmem培养基中孵育。孵育24h后,分别加入不同浓度的阿霉素溶液或载药共聚物胶束溶液,并保持dox绝对终浓度分别为0、0.1、0.2、0.3、0.5、1.0、2.0和3.0μg/ml。继续孵育24h后,每孔加入10μl cck-8溶液,孵育1h,轻轻振荡后,在酶标仪(biotek,elx800,usa)上测定λ=490nm处的吸光度,以λ=630nm为参比,按公式2计算细胞存活率,并以6个复孔的平均值作为最终结果。
[0067]
相对细胞存活率=(od
490(sample)-od
630(sample)
)/(od
490(control)-od
630(control)
)
×
100%
ꢀꢀ
公式2
[0068]
由图6可见,随着载药共聚物胶束浓度的增加,mcf-7细胞存活率迅速下降,当负载药物的浓度达到2.0和3.0μg/ml时,细胞相对存活率分别为26.9%和5.7%,与游离阿霉素的19.6%和5.7%相当,表明制备所得载药共聚物胶束能够很好的抑制肿瘤细胞的增殖,可以作为高效药物输送载体使用。
[0069]
以上所述仅为本发明的优选实施方式,并不用于限制本发明。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips