
 商标分类
商标分类  商标转让
商标转让 
一种自模板调控的C/CoS2纳米管结构制备方法与流程
 2021-01-31 00:01:48|
2021-01-31 00:01:48| 368|
368| 起点商标网
起点商标网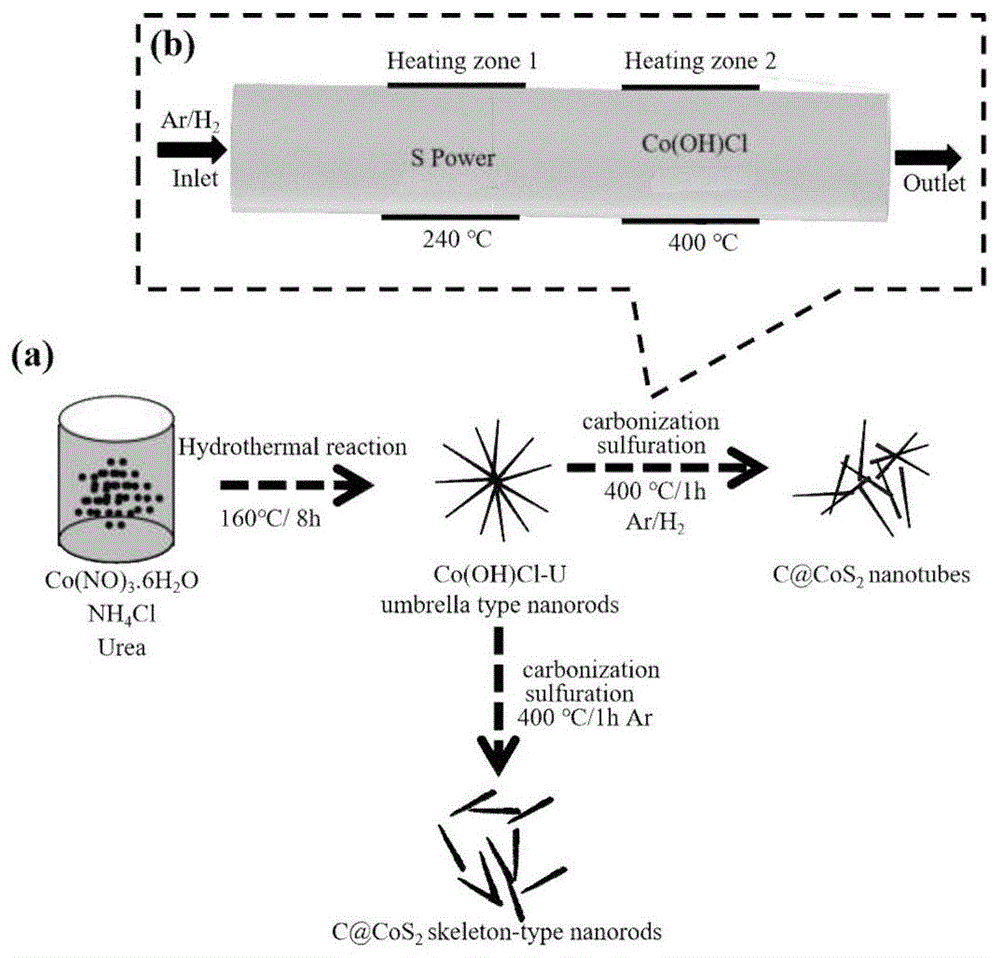
本发明涉及结构可控的c/cos2纳米管制备方法,属于纳米光电子材料技术领域。
背景技术:
全球能源危机已成为严重的世界性问题。石油、煤炭、天然气等传统能源是一次能源,不可再生,寻求可持续、可再生、清洁能源显得尤为重要。在过去的几十年里,人们致力于开发氢燃料、太阳能电池、锂硫电池、金属空气电池、二氧化碳电化学还原、钙钛矿太阳能电池、传感器等新能源,其中氢能源则成为科研学者最为关注的研究对象。众所周知,氢一直被认为是一种清洁、地球资源丰富、环境友好的能源。但是,若要开发出足够的氢能,则需要提供较大偏压,这对设备功率提出很高的要求。那么,能否提出一种改进方式,以降低电耗为出发点,实现高效催化的目的。迄今为止,众多科研人员都在极力寻找高效制氢的催化剂,如贵金属pt和pd则成为研究电催化性能的首选材料。然而,这些贵金属含量少,价格昂贵,从长远角度考虑,它并不是一种合适的催化剂,因此,找到一种高效,无污染且成本低的催化剂来代替贵金属显得尤为重要。
近年来,过渡金属硫属化合物mx2,其中m通常为mo,w和co等;x通常为s,se等,由于独特的电化学特性,他们被认为是最有可能取代贵金属的硫属化合物。其中,mos2是最为典型的催化剂之一,它在电催化领域的应用较为广泛。然而,纯制备的mos2存在一些缺点,如电导率低,催化活性位点少,这就限制它在催化领域的进一步发展。与mos2不同,cos2同样具有超高电导率以及极好催化性能,其得到研究者的追捧。并且它的制备工艺简单,产物纯度高,易于控制,可以得到不同形貌产物,如纳米管、纳米线、纳米片、纳米空心立方体、纳米花和纳米颗粒等。此外,结构调控也是增强电化学性能的重要因素,如添加碳基导电材料、构建核-壳复合结构、构建异质结构以及掺杂n,se于纳米材料等方法。然而,上述研究均存在一些弊端,主要有制备方法难度大,产物结构不可控,催化剂活性位点少等问题,从而影响电极材料的电化学性能。
技术实现要素:
本发明是为避免上述现有技术所存在的不足之处,提出一种简便自模板调控的c/cos2纳米管结构制备方法,旨在解决现有cos2比表面积小、化学活性位少、电催化性能低等问题,同时提高该电极材料在电及光电催化方面的应用。
本发明解决技术问题采用如下技术方案:
一种自模板调控的c/cos2纳米管结构制备方法,包括如下步骤:
a、称取0.58gco(no3)2·6h2o或者0.47gcocl2·6h2o、0.27gnh4cl、0.6gurea,将其混合均匀放到50ml烧杯中,加入35ml去离子水,使其搅拌均匀,然后放到50ml反应釜中,置于烘箱中反应6-12小时,反应温度为80-180℃,得到初始产物co(oh)cl-urea;
b、待反应冷却后,取出初始产物co(oh)cl-urea,进而对初始产物co(oh)cl-urea进行超声以保证其纯度,超声时间20分钟,再使用酒精和去离子水多次洗涤初始产物co(oh)cl-urea,最后将初始产物co(oh)cl-urea置于真空干燥箱里烘干12-24小时,烘箱温度为60℃;
c、称量0.01g初始产物co(oh)cl-urea置于化学气相沉积系统中的高温区,温度设定为400℃;将0.1gs粉置于化学气相沉积系统中的低温区,温度设定为240℃;初始产物co(oh)cl-urea与s粉之间的位置保持在15-20cm;整个化学气相反应在ar/h2气体保护下进行,反应时间为1小时,待反应冷却后得到c/cos2。
作为本发明的进一步改进,所述步骤a中反应温度为140℃,所述步骤c中ar/h2比为100%:0%。
作为本发明的进一步改进,所述步骤a中反应温度为140℃,所述步骤c中ar/h2比为99%:1%。
作为本发明的进一步改进,所述步骤a中反应温度为140℃,所述步骤c中ar/h2比为95%:5%。
作为本发明的进一步改进,所述步骤a中反应温度为140℃,所述步骤c中ar/h2比为90%:10%。
与现有技术相比,本发明的有益效果体现在:
1.制备方法简单,产物纯度高;
2.产物形貌可控。通过控制反应ar/h2气体比,可以得到不同形貌;
3.制备的产物活性位点多。纳米管的比表面积大,催化活性位点多,有利于催化性能提高;
4.本发明方法具有普适性。同样适用于其他硫属纳米材料在电存储及电催化等方面的应用。
附图说明
图1a为实施例1、2、3和4中的工艺流程图;
图1b为实施例1、2、3和4中的化学气相工艺流程图;
图2为不同温度下前驱物co(oh)cl-urea的扫描电子显微镜图;
图2a-b为80℃下的前驱物co(oh)cl-urea的形貌图;
图2c-d为100℃下的前驱物co(oh)cl-urea的形貌图;
图2e-f为140℃下的前驱物co(oh)cl-urea的形貌图;
图2g-h为140℃下的前驱物co(oh)cl-urea的形貌图;
图2i-j为180℃下的前驱物co(oh)cl-urea的形貌图;
图3为实施例1、2、3和4样品的形成机理图和不同ar/h2比下的扫描电子显微镜图;
图4为实施例1、2、3和4不同温度下的硫化产物的扫描电子显微镜图;
图4a-c为300℃下硫化产物的扫描电子显微镜图;
图4d-f为350℃下硫化产物的扫描电子显微镜图;
图4g-i为400℃下硫化产物的扫描电子显微镜图;
图4j-l为600℃下硫化产物的扫描电子显微镜图;
图4m-o为800℃下硫化产物的扫描电子显微镜图;
图5a-b为实施例3样品的扫描电子显微镜;
图5c-d为实施例3样品的透射电子显微镜;
图5e-f为实施例3样品的电子能谱图;
图6a-c为本发明在实施例3样品的光电子能谱图;
图6d为本发明在实施例3样品的热重分析图。
具体实施方式
下面结合具体实施例和附图对本发明做进一步说明,而不限制于本发明的范围。
a、称取0.58gco(no3)2·6h2o或者0.47gcocl2·6h2o、0.27gnh4cl、0.6gurea,将其混合均匀放到50ml烧杯中,加入35ml去离子水,使其搅拌均匀,然后放到50ml反应釜中,置于烘箱中反应6-12小时,反应温度为80-180℃,得到初始产物co(oh)cl-urea;
b、待反应冷却后,取出初始产物co(oh)cl-urea,进而对初始产物co(oh)cl-urea进行超声以保证其纯度,超声时间20分钟,再使用酒精和去离子水多次洗涤初始产物co(oh)cl-urea,最后将初始产物co(oh)cl-urea置于真空干燥箱里烘干12-24小时,烘箱温度为60℃;
c、称量0.01g初始产物co(oh)cl-urea置于化学气相沉积系统中的高温区,温度设定为400℃;将0.1gs粉置于化学气相沉积系统中的低温区,温度设定为240℃;初始产物co(oh)cl-urea与s粉之间的位置保持在15-20cm;整个化学气相反应在ar/h2气体保护下进行,反应时间为1小时,待反应冷却后得到c/cos2。
作为本发明的进一步改进,所述步骤a中反应温度为140℃,所述步骤c中ar/h2比为100%:0%。
作为本发明的进一步改进,所述步骤a中反应温度为140℃,所述步骤c中ar/h2比为99%:1%。
作为本发明的进一步改进,所述步骤a中反应温度为140℃,所述步骤c中ar/h2比为95%:5%。
作为本发明的进一步改进,所述步骤a中反应温度为140℃,所述步骤c中ar/h2比为90%:10%。
实施例1
本实施例按如下步骤制备c/cos2链式纳米棒:
a、称取0.58gco(no3)2·6h2o或者0.47gcocl2·6h2o、0.27gnh4cl、0.6gurea,将其混合均匀放到50ml烧杯中,加入35ml去离子水,使其搅拌均匀,然后放到50ml反应釜中,置于烘箱中反应6-12小时,反应温度为140℃,得到初始产物co(oh)cl-urea;
b、待反应冷却后,取出初始产物co(oh)cl-urea,进而对初始产物co(oh)cl-urea进行超声以保证其纯度,超声时间20分钟,再使用酒精和去离子水多次洗涤初始产物co(oh)cl-urea,最后将初始产物co(oh)cl-urea置于真空干燥箱里烘干12-24小时,烘箱温度为60℃;
c、称量0.01g初始产物co(oh)cl-urea置于化学气相沉积系统中的高温区,温度设定为400℃;将0.1gs粉置于化学气相沉积系统中的低温区,温度设定为240℃;初始产物co(oh)cl-urea与s粉之间的位置保持在15-20cm;整个化学气相反应在ar/h2气体保护下进行,其中ar/h2比为100%:0%,反应时间为1小时,待反应冷却后得到c/cos2。
实施例2
本实施例按如下步骤制备c/cos2纳米棒和纳米管的混相:
a、称取0.58gco(no3)2·6h2o或者0.47gcocl2·6h2o、0.27gnh4cl、0.6gurea,将其混合均匀放到50ml烧杯中,加入35ml去离子水,使其搅拌均匀,然后放到50ml反应釜中,置于烘箱中反应6-12小时,反应温度为140℃,得到初始产物co(oh)cl-urea;
b、待反应冷却后,取出初始产物co(oh)cl-urea,进而对初始产物co(oh)cl-urea进行超声以保证其纯度,超声时间20分钟,再使用酒精和去离子水多次洗涤初始产物co(oh)cl-urea,最后将初始产物co(oh)cl-urea置于真空干燥箱里烘干12-24小时,烘箱温度为60℃;
c、称量0.01g初始产物co(oh)cl-urea置于化学气相沉积系统中的高温区,温度设定为400℃;将0.1gs粉置于化学气相沉积系统中的低温区,温度设定为240℃;初始产物co(oh)cl-urea与s粉之间的位置保持在15-20cm;整个化学气相反应在ar/h2气体保护下进行,其中ar/h2比为99%:1%,反应时间为1小时,待反应冷却后得到c/cos2。
实施例3
本实施例按如下步骤制备c/cos2纳米管
a、称取0.58gco(no3)2·6h2o或者0.47gcocl2·6h2o、0.27gnh4cl、0.6gurea,将其混合均匀放到50ml烧杯中,加入35ml去离子水,使其搅拌均匀,然后放到50ml反应釜中,置于烘箱中反应6-12小时,反应温度为140℃,得到初始产物co(oh)cl-urea;
b、待反应冷却后,取出初始产物co(oh)cl-urea,进而对初始产物co(oh)cl-urea进行超声以保证其纯度,超声时间20分钟,再使用酒精和去离子水多次洗涤初始产物co(oh)cl-urea,最后将初始产物co(oh)cl-urea置于真空干燥箱里烘干12-24小时,烘箱温度为60℃;
c、称量0.01g初始产物co(oh)cl-urea置于化学气相沉积系统中的高温区,温度设定为400℃;将0.1gs粉置于化学气相沉积系统中的低温区,温度设定为240℃;初始产物co(oh)cl-urea与s粉之间的位置保持在15-20cm;整个化学气相反应在ar/h2气体保护下进行,其中ar/h2比为95%:5%,反应时间为1小时,待反应冷却后得到c/cos2。
实施例4
本实施例按如下步骤制备c/cos2纳米管
a、称取0.58gco(no3)2·6h2o或者0.47gcocl2·6h2o、0.27gnh4cl、0.6gurea,将其混合均匀放到50ml烧杯中,加入35ml去离子水,使其搅拌均匀,然后放到50ml反应釜中,置于烘箱中反应6-12小时,反应温度为140℃,得到初始产物co(oh)cl-urea;
b、待反应冷却后,取出初始产物co(oh)cl-urea,进而对初始产物co(oh)cl-urea进行超声以保证其纯度,超声时间20分钟,再使用酒精和去离子水多次洗涤初始产物co(oh)cl-urea,最后将初始产物co(oh)cl-urea置于真空干燥箱里烘干12-24小时,烘箱温度为60℃;
c、称量0.01g初始产物co(oh)cl-urea置于化学气相沉积系统中的高温区,温度设定为400℃;将0.1gs粉置于化学气相沉积系统中的低温区,温度设定为240℃;初始产物co(oh)cl-urea与s粉之间的位置保持在15-20cm;整个化学气相反应在ar/h2气体保护下进行,其中ar/h2比为90%:10%,反应时间为1小时,待反应冷却后得到c/cos2。
图1是实施例1、2、3和4产物的工艺流程图。其中图1a为实施例1、2、3和4中的工艺流程图;图1b为实施例1、2、3和4中的化学气相工艺流程图。图中明显可以看出,第一步采用低温溶剂热法制备前驱体co(oh)cl-urea;第二步采用化学气相沉积法制备出c@cos2产物,通过改变ar/h2气体比,可得到不同产物。
图2为不同温度下前驱物co(oh)cl-urea的扫描电子显微镜图。从图2a-b中可以看出,溶剂热反应温度设定80℃,产物出现少量纳米棒;当温度升高至100℃时,如图2c-d所示,产物出现部分无序纳米棒,且有聚集现象;进一步升高温度至140℃,如图2e-f,此刻产物形貌发生较大改变,形成伞型纳米棒结构;当反应温度达到160℃,如图2g-h,产物形貌呈现伞状纳米棒结构;继续升温至180℃,如图2i-j所示,产物形貌不再发生变化。
图3为实施例1、2、3和4样品的形成机理图和不同ar/h2气体比的扫描电子显微镜图;图3a为产物形成机理图。图3b是纯ar(100%ar)气氛下的化学气相沉积产物,呈现骨架型纳米棒。当h2加入量为1%(1%h2,99%ar)时,产物变为棒管混合结构,如图3c所示。随着h2用量进一步增加,产物中管状结构越来越明显。当h2量达到5%(5%h2,95%ar)时,大部分纳米棒转化为纳米管,如图3d所示。继续增加h2量,当ar/h2比超过10%(10%h2,90%ar)时,如图3e所示,纳米管表面粗糙,呈现破坏样貌。综上所述,气体环境在纳米棒或纳米管合成中起着决定性作用。即当氢离子浓度较低时,其电子传递不会改变co(oh)cl-urea针状纳米棒表面性质,不会引起内部反应。随着氢浓度增加,其电子转移足以改变co(oh)cl-urea表面性质,促进其内部反应。随着co2+、oh-和尿素分子向外扩散,反应进一步推进,由于固体扩散活化能远大于反应活化能,形成空心管状结构。
图4为实施例1、2、3和4不同温度下硫化产物的扫描电子显微镜图;图4a-c为300℃下硫化产物扫描电子显微镜图;图4d-f为400℃下硫化产物扫描电子显微镜图;图4g-i为500℃下硫化产物的扫描电子显微镜图;图4j-l为600℃下硫化产物的扫描电子显微镜图;图4m-o为800℃下硫化产物的扫描电子显微镜图;从图中可以看出,反应温度在400℃时的产物,纳米管结构最为明显,由此得出,反应最优温度为400℃。
图5a-b为实施例3样品的扫描电子显微镜;从图中可以看出产物呈现中空结构。图5c-d为实施例3样品的透射电子显微镜。如图5c是产物tem图,从图中可以看出,该结构呈管状,从高分辨率tem(hrtem)图像可以看出,晶体的晶格条纹明显,其晶格间距也与文献报道的cos2的值一致,进一步证明产物的存在。图5f能谱测试结果显示co与s比为1:2,进一步证实产物中含有cos2。
图6a-c为本发明在实施例3样品的光电子能谱图;图6d为本发明在实施例3样品的热重分析图。图中分析可知,位于778.3ev和780.5ev处的峰分别对应于co2p3/2和co2p1/2,这与文献报道的cos2中的co2p值十分吻合。此外,两个位于785.9和802.5ev的xps峰分别对应于coox中的co2p3/2和co2p1/2,这个结果证实了coox的存在。此外,可以发现,s2pxps谱,如图6b所示,位于162.7ev,164.2ev分别对应于s2p3/2和s2p1/2,这意味着s2-的存在。同时,为进一步证明产物中碳的存在,也测试了c1s光谱,如图6c所示,从图中可以看出,该谱存在非氧合碳(c-c284.9ev)和碳(c-o287ev)两个特征峰,且c的存在形式可能是无定形的。为进一步证明碳含量,如图6d所示,采用热重分析法(tga)测定cos2中碳含量。经计算可得非晶碳含量约为22.3%(wt%)。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



