
 商标分类
商标分类  商标转让
商标转让 
化合物单晶及其制备方法与流程
 2021-01-30 19:01:50|
2021-01-30 19:01:50| 419|
419| 起点商标网
起点商标网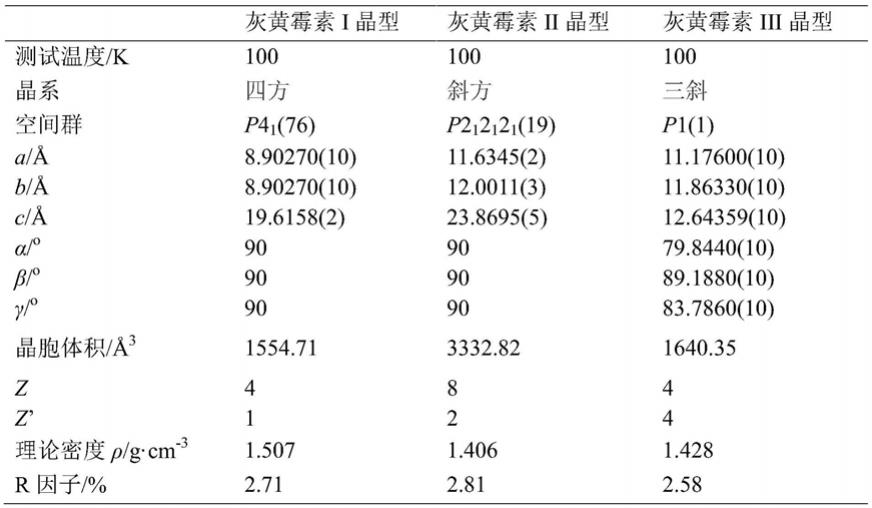
[0001]
本发明涉及单晶结构解析领域,特别是涉及化合物单晶及其制备方法。
背景技术:
[0002]
单晶结构解析对于阐明化合物空间构象、化学反应机理、药物与靶点结合机制等超分子作用过程具有重要意义。目前,单晶结构解析多采用单晶x射线衍射分析技术。单晶结构解析的常规步骤包括:单晶培养,单晶测试和数据解析。培养尺寸大、质量好的单晶往往是限速步骤。
[0003]
传统培养单晶的方法基于溶液结晶,包括缓慢挥发法、溶剂转移法等,这些方法变量多(需要尝试不同溶剂、浓度、温度、挥发速度等)、实验周期长。多晶型是化合物常见的现象,溶液法难以选择性培养多晶型化合物目标晶型的单晶。
[0004]
熔体结晶是用来培养单晶硅等无机材料的常规方法,通常包括提拉法、泡生法、坩埚下降法等,需要特殊装置,且必须引入籽晶,适合制备用于工业的大尺寸单晶材料,而无法用于有机合成、药物研发等领域涉及到的单晶培养,尤其是有机化合物单晶培养。常规化合物熔体结晶通常获得多晶,难以获得单晶,其原因在于化合物从熔体中自发成核需要较高的过冷度(过冷度=熔融温度-结晶温度)、而较高的过冷度下晶体通常会由于生长前沿的二次成核而形成多晶(通常为球晶)。只有少数特殊晶型例外,如灰黄霉素ii晶型(chemical communications 2018,54(4):358-361)可在高过冷度下形成具有规则外形的单晶,硝苯地平β晶型(crystal growth&design 2012,12(4):2037-2043),在球晶中会形成较大区域的单晶形貌。
[0005]
近来学者们利用熔体结晶方法发现一些药用化合物的新晶型,这些晶型通常只能够由熔体结晶获得。由于前述原因,这些晶型的单晶难以培养,因此单晶结构很难解析。例如硝苯地平γ晶型1977年首次由熔体结晶获得(archiv der pharmazie 1977,310(2):116-118),42年间关于它的单晶结构研究从未停止过,但一直无法解析。再如2013年首次报道的灰黄霉素iii晶型(journal of pharmaceutical sciences 2013,102(2):462-468)和2016年报道的维罗非尼β晶型和γ晶型(crystal growth&design 2016,16(10):6033-6042),单晶结构也一直无法解析。
技术实现要素:
[0006]
基于此,本发明提供一种化合物单晶的制备方法,从过冷熔体中选择性培养出化合物目标晶型的大尺寸、高质量单晶,成功解析了多种业界多年来无法解析的单晶结构。同时,实验结果表明,该单晶制备方法可以从原料药出发,快速地制备手性药物的单晶、解析单晶结构、确定绝对构型。
[0007]
具体技术方案为:
[0008]
一种化合物单晶的制备方法,包括以下步骤:
[0009]
1)熔融化合物,获取过冷熔体,向所述过冷熔体中引入化合物目标晶型的晶种,或
使所述过冷熔体自发成核,得化合物目标晶型的多晶样品;若所述化合物本身即为目标晶型的多晶样品,则直接执行步骤2);
[0010]
2)以0.1-100℃/min的升温速率第一次升温,将所述化合物目标晶型的多晶样品升温至t
3
,所述t
3
比t
m
低0.5-8℃,然后以0.1-20℃/min的升温速率第二次升温,至获得1-10颗化合物目标晶型的单晶晶核,若所有晶体完全熔融,仍没有获得1-10颗化合物目标晶型的单晶晶核,则重复步骤1)和步骤2),调整所述第二次升温的升温速率,直至获得1-10颗化合物目标晶型的单晶晶核,所述t
m
为所述化合物目标晶型的熔点;
[0011]
3)降温培养,得单晶。
[0012]
在其中一个实施例中,所述第二次升温的升温速率为0.1-10℃/min。
[0013]
在其中一个实施例中,所述第二次升温的升温速率为0.1-1℃/min。
[0014]
在其中一个实施例中,所述t
3
比t
m
低0.5-3℃。
[0015]
在其中一个实施例中,所述步骤3)中,降温至t
4
,所述t
4
>t
m-0.1(t
m
+273℃)。
[0016]
在其中一个实施例中,所述步骤3)包括:
[0017]
a)将所述1-10颗化合物目标晶型的单晶晶核降温至温度t
4
,恒温培养;
[0018]
b)若未出现分叉生长,即得单晶;若出现分叉,则重复步骤2),至获得1-10颗化合物目标晶型单晶晶核,降温至温度t
5
,t
4
<t
5
<t
m
;在所述温度t
5
下,恒温培养,获得单晶。
[0019]
在其中一个实施例中,步骤1)中,在温度t
1
下向所述过冷熔体中引入化合物目标晶型的晶种,或者在温度t
2
下使所述过冷熔体自发成核,得所述化合物目标晶型的多晶样品,其中,所述t
1
和t
2
均高于t
g
、且均低于t
m
,所述t
g
为所述化合物的玻璃化转变温度。
[0020]
在其中一个实施例中,在温度t
1
下向所述过冷熔体中引入化合物目标晶型的晶种,得化合物目标晶型的多晶样品,其中,t
1
>t
g
,所述t
g
为所述化合物的玻璃化转变温度。
[0021]
本发明还提供一种由上述制备方法制得的化合物单晶。
[0022]
在其中一个实施例中,所得化合物单晶为灰黄霉素i晶型单晶、灰黄霉素ii晶型单晶、灰黄霉素iii晶型单晶、维罗非尼β晶型单晶、维罗非尼γ晶型单晶、硝苯地平α晶型单晶、硝苯地平β晶型单晶、硝苯地平γ晶型单晶、烟酰胺α晶型单晶、烟酰胺β晶型单晶、烟酰胺γ晶型单晶、烟酰胺δ晶型单晶、烟酰胺ε晶型单晶、烟酰胺ζ晶型单晶或r-克唑替尼α晶型单晶。
[0023]
使用x射线单晶衍射仪测定上述单晶的晶体结构,得到各晶型的单晶结构,如下所示:
[0024]
本发明所述的灰黄霉素i晶型为四方晶系,空间群为p4
1
(76),晶轴为(76),晶轴为晶面间夹角α=90
°
,β=90
°
,γ=90
°
。
[0025]
本发明所述的灰黄霉素ii晶型为斜方晶系,空间群为p2
1
2
1
2
1
(19),晶轴为(19),晶轴为晶面间夹角α=90
°
,β=90
°
,γ=90
°
。
[0026]
本发明所述的灰黄霉素iii晶型为三斜晶系,空间群为p1(1),晶轴为本发明所述的灰黄霉素iii晶型为三斜晶系,空间群为p1(1),晶轴为晶面间夹角α=79.8440(10)
°
,β=89.1880(10)
°
,γ=83.7860(10)
°
。
[0027]
本发明所述的维罗非尼β晶型为单斜晶系,空间群为p2
1
/c(14),晶轴为/c(14),晶轴为晶面间夹角α=90
°
,β=94.9460(10)
°
,
γ=90。
[0028]
本发明所述的维罗非尼γ晶型为三斜晶系,空间群为晶轴为晶轴为晶面间夹角α=98.5010(10)
°
,β=100.5350(10)
°
,γ=105.5980(10)
°
。
[0029]
本发明所述的硝苯地平α晶型为单斜晶系,空间群为p2
1
/c(14),晶轴为/c(14),晶轴为晶面间夹角α=90
°
,β=94.879(3),γ=90
°
。
[0030]
本发明所述的硝苯地平β晶型为三斜晶系,空间群为晶轴为晶轴为晶面间夹角α=61.203(2)
°
,β=79.578(2)
°
,γ=81.873(2)
°
。
[0031]
本发明所述的硝苯地平γ晶型单晶为单斜晶系,空间群为p2
1
/c(14),其晶轴为晶面间夹角α=90
°
,β=108.858(4)
°
,γ=90
°
。本发明所述的烟酰胺α晶型为单斜晶系,空间群为p2
1
/c(14),晶轴为/c(14),晶轴为晶面间夹角α=90
°
,β=98.394(4)
°
,γ=90
°
。
[0032]
本发明所述的烟酰胺β晶型为单斜晶系,空间群为p2/n(13),晶轴为本发明所述的烟酰胺β晶型为单斜晶系,空间群为p2/n(13),晶轴为晶面间夹角α=90(10)
°
,β=101.955(2)
°
,γ=90
°
。
[0033]
本发明所述的烟酰胺γ晶型为单斜晶系,空间群为p2
1
/c(14),晶轴为/c(14),晶轴为晶面间夹角α=90
°
,β=104.650(5)
°
,γ=90
°
。
[0034]
本发明所述的烟酰胺δ晶型为单斜晶系,空间群为p2
1
(4),晶轴为(4),晶轴为晶面间夹角α=90
°
,β=94.2560(10)
°
,γ=90
°
。
[0035]
本发明所述的烟酰胺ε晶型为三斜晶系,空间群为晶轴为晶轴为晶面间夹角α=71.499(5)
°
,β=85.676(6)
°
,γ=85.202(5)
°
。
[0036]
本发明所述的烟酰胺ζ晶型为单斜晶系,空间群为p2
1
(4),晶轴为(4),晶轴为晶面间夹角α=90
°
,β=102.5800(10)
°
,γ=90
°
。
[0037]
本发明所述的r-克唑替尼α晶型为单斜晶系,空间群为c2(5),晶轴为克唑替尼α晶型为单斜晶系,空间群为c2(5),晶轴为晶面间夹角α=90
°
,β=100.491(3)
°
,γ=90
°
。
[0038]
本发明的有益效果如下:
[0039]
采用本发明所述的方法,可以用极少量的样品(微克级)从过冷熔体中快速获得大
尺寸、高质量的化合物单晶。成功解析了多种业界多年来无法解析的单晶结构,包括维罗非尼β晶型和γ晶型、硝苯地平γ晶型、灰黄霉素iii晶型。总体思想在于,通过在温度t
1
下引入化合物目标晶型晶种或者在温度t
2
下自发成核,获得化合物目标晶型的多晶样品,再以0.1-100℃/min的升温速率将所述化合物目标晶型的多晶样品升温至t
3
,所述t
3
比t
m
低0.5-8℃,然后再以0.1-20℃/min的升温速率第二次升温,通过精细控温,熔融大部分晶体,获得含有1-10颗化合物目标晶型单晶晶核的熔体,然后将所述熔体降温培养,获得单晶。该方法无需使用溶剂,可对单晶培养过程进行实时可视化监控。
[0040]
上述方法适用于熔融时受热不分解或分解不严重的化合物以及熔融时不升华或升华不严重的化合物。该方法适用于只能够通过熔体结晶获得的晶型或难以从溶液中培养单晶的化合物,也适用于能够通过溶液法培养单晶的化合物。该方法只需要微克级样品,变量少,效率高,可控性强,且能够选择性培养化合物目标晶型的单晶。
附图说明
[0041]
图1为实施例1的单晶制备过程中各阶段的显微照片;
[0042]
图2为实施例1制备的灰黄霉素i晶型单晶、实施例2制备的灰黄霉素ii晶型单晶和实施例3制备的灰黄霉素iii晶型单晶的显微照片;
[0043]
图3为实施例1中灰黄霉素i晶型粉末x射线衍射谱图、实施例2中灰黄霉素ii晶型粉末x射线衍射谱图和实施例3中灰黄霉素iii晶型粉末x射线衍射谱图;其中,标识experimental的谱图为实验测得的粉末x射线衍射谱图,标识calculated的谱图为单晶结构拟合所得的粉末x射线衍射谱图;
[0044]
图4为实施例1制备的灰黄霉素i晶型单晶晶胞堆积图、实施例2制备的灰黄霉素ii晶型单晶晶胞堆积图和实施例3制备的灰黄霉素iii晶型单晶晶胞堆积图;
[0045]
图5为实施例4和实施例5中维罗非尼α、β和γ三种晶型多晶共生样品显微照片;
[0046]
图6为实施例4制备的维罗非尼β晶型单晶和实施例5制备的维罗非尼γ晶型单晶的显微照片;
[0047]
图7为实施例4中维罗非尼β晶型粉末x射线衍射谱图和实施例5中维罗非尼γ晶型粉末x射线衍射谱图;其中,标识experimental的谱图为实验测得的粉末x射线衍射谱图,标识calculated的谱图为单晶结构拟合所得的粉末x射线衍射谱图;
[0048]
图8为实施例4制备的维罗非尼β晶型的单晶晶胞堆积图和实施例5制备的维罗非尼γ晶型的单晶晶胞堆积图;
[0049]
图9为实施例6制备的硝苯地平α晶型单晶、实施例7制备的硝苯地平β晶型单晶和实施例8制备的硝苯地平γ晶型单晶的显微照片;
[0050]
图10为实施例6中硝苯地平α晶型粉末x射线衍射谱图;其中,标识experimental的谱图为实验测得的粉末x射线衍射谱图,标识calculated的谱图为单晶结构拟合所得的粉末x射线衍射谱图;
[0051]
图11为实施例6制备的硝苯地平α晶型单晶晶胞堆积图、实施例7制备的硝苯地平β晶型单晶晶胞堆积图和实施例8制备的硝苯地平γ晶型单晶晶胞堆积图;
[0052]
图12为实施例9制备的烟酰胺α晶型单晶、实施例10制备的烟酰胺β晶型单晶、实施例11制备的烟酰胺γ晶型单晶、实施例12制备的烟酰胺δ晶型单晶、实施例13制备的烟酰胺
ε晶型单晶和实施例14制备的烟酰胺ζ晶型单晶的显微照片;
[0053]
图13为实施例9中烟酰胺α晶型粉末x射线衍射谱图、实施例10中烟酰胺β晶型粉末x射线衍射谱图、实施例11中烟酰胺γ晶型粉末x射线衍射谱图、实施例12中烟酰胺δ晶型粉末x射线衍射谱图、实施例13中烟酰胺ε晶型粉末x射线衍射谱图和实施例14中烟酰胺ζ晶型粉末x射线衍射谱图;其中,标识experimental的谱图为实验测得的粉末x射线衍射谱图,标识calculated的谱图为单晶结构拟合所得的粉末x射线衍射谱图;
[0054]
图14为实施例9制备的烟酰胺α晶型单晶晶胞堆积图、实施例10制备的烟酰胺β晶型单晶晶胞堆积图、实施例11制备的烟酰胺γ晶型单晶晶胞堆积图、实施例12制备的烟酰胺δ晶型单晶晶胞堆积图、实施例13制备的烟酰胺ε晶型单晶晶胞堆积图和实施例14制备的烟酰胺ζ晶型单晶晶胞堆积图;
[0055]
图15为实施例15制备的r-克唑替尼α晶型单晶的显微照片;
[0056]
图16为实施例15中r-克唑替尼α晶型粉末x射线衍射谱图;其中,标识experimental的谱图为实验测得的粉末x射线衍射谱图,标识calculated的谱图为单晶结构拟合所得的粉末x射线衍射谱图;
[0057]
图17为实施例15制备的r-克唑替尼α晶型单晶晶胞堆积图。
具体实施方式
[0058]
以下结合具体实施例对本发明的化合物单晶及其制备方法作进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。
[0059]
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。
[0060]
本专利中所使用的所有温度均为摄氏温度。本专利中所述的多晶样品,其结晶度(结晶部分占总样品的比例)可为1%-100%。
[0061]
结晶是受热力学和动力学共同控制的过程,并且具有化合物依赖性。化合物在熔点以上会熔融形成熔体(液态),熔体降温至熔点以下,可能出现两种现象:其一,析出晶体;其二,保持非晶态。处于熔点和玻璃化转变温度之间的非晶态,称为过冷液体(supercooled liquids)或者过冷熔体(undercooled melts);温度低于玻璃化转变温度时,非晶态称为玻璃态(glass)。过冷熔体比玻璃态具有更高的分子活动能力,因此更容易发生分子聚集、自组装成为单晶晶核、生长为晶体。本发明正是基于化合物的过冷熔体实现单晶的高效培养。
[0062]
结晶包括成核和晶体生长两个步骤。这两个步骤对过冷度(熔点与实验温度之差)的依赖性不同。低过冷度下,过冷熔体粘度小、分子活动性高、结晶驱动力小,不易成核,但晶体生长速率很快(过冷度非常低时晶体生长速率会急速减慢);高过冷度下,过冷熔体粘度大、分子活动性低,晶体生长慢,但结晶驱动力大,因此容易成核,而晶体生长过程中晶体前沿的二次成核通常导致球晶的形成,球晶被认为是多个单晶的聚集体。
[0063]
基于上述原理,本发明通过部分熔融化合物目标晶型的多晶样品,然后降温,在过冷熔体中原位形成化合物目标晶型的低密度单晶晶核,在低过冷度下恒温培养,成功获得
单晶。本申请的发明人在实验过程中发现,在化合物熔点附近,控制升温速率,可获得只含有1-10颗单晶晶核的熔体,此时,选择合适的温度t
4
[t
m-t
4
<0.1(t
m
+273℃)]对其进行培养,可获得单晶。其中,升温的速率的控制,对成功获得1-10颗单晶晶核至关重要;培养温度的选择,对于培养单晶至关重要。
[0064]
获得含有化合物目标晶型单晶晶核的熔体,包含4种情况:其一,当原料多晶样品只含有目标晶型时,可将原料多晶样品直接升温至目标晶型的熔点附近,获得目标熔体;其二,当原料多晶样品中含有目标晶型和熔点较低的其他晶型时,可直接升温至目标晶型的熔点附近,获得目标熔体;其三,当原料多晶样品中含有目标晶型和熔点较高的其他晶型时,需要先制备仅含有目标晶型的多晶样品,或者含有目标晶型和熔点较低晶型的多晶样品,再升温至目标晶型的熔点附近,获得目标熔体;其四,当原料多晶样品中不含目标晶型时,需要先制备仅含有目标晶型的多晶样品,或者含有目标晶型和熔点较低晶型的多晶样品,再升温至目标晶型的熔点附近,获得目标熔体。
[0065]
制备仅含有目标晶型的多晶样品,或者含有目标晶型和熔点较低晶型的多晶样品,包含2种情况:其一,将多晶样品完全熔融,降温至合适的温度,使过冷熔体自发成核、析出晶体;其二,如果自发成核难以获得仅含有目标晶型的多晶样品或者含有目标晶型和熔点较低晶型的多晶样品时,需要在合适的过冷度下、通过引入目标晶型晶种的方法,制备仅含有目标晶型的多晶样品,或者含有目标晶型和熔点较低晶型的多晶样品。
[0066]
本发明针对具有不同特征的化合物体系有不同的处理策略。具体如下:
[0067]
灰黄霉素的分子结构如下所示:
[0068][0069]
实施例1
[0070]
本实施例提供一种灰黄霉素i晶型单晶的制备方法,步骤如下(如图1所示):
[0071]
步骤1)使用的仪器为热台-偏光显微镜,linkam热台位于显微镜载物台上,热台的中间为样品放置台,将少许灰黄霉素粉末状放置于玻片上,再置于样品放置台(linkam热台控温精度为
±
0.1℃,由其配套软件控制)。
[0072]
步骤2)将linkam热台以100℃/min升温到218℃,再以10℃/min缓慢升温至220℃,再以1℃/min缓慢升温至221℃(该温度为灰黄霉素i晶型的熔点)附近。
[0073]
步骤3)若观察到1-10颗灰黄霉素i晶型的单晶晶核,则以150℃/min降温速率降温至215℃,恒温培养,让所述1-10颗单晶晶核生长至可供单晶衍射测试的尺寸,得灰黄霉素i晶型单晶。
[0074]
步骤4)若所有晶体完全熔融,仍没有获得1-10颗灰黄霉素i晶型的单晶晶核,则重复步骤1)和步骤2),升温到220℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1-10颗灰黄霉素i晶型的单晶晶核,执行步骤3)。
[0075]
本实施例所制备的灰黄霉素i晶型单晶的显微照片如图2(1)所示。
[0076]
对灰黄霉素i晶型粉末样品进行粉末x射线衍射(pxrd)测定,使用rigaku d-max/2200vpc粉末衍射仪进行常温测试,测试条件为:以cu kα为光源,波长为电压40kv,电流26ma,扫描速度6
°
/min,扫描范围5-45
°
(2θ)。测试谱图如图3(1)所示。
[0077]
对所制备的灰黄霉素i晶型单晶进行x射线单晶衍射测试。使用日本理学x射线单晶衍射仪(型号xtalab synergy)收集单晶衍射数据,测试条件为:温度100k电压40kv,电流30ma,cu kα射线,波长为利用crysalispro 1.171.39.46软件进行单晶数据收集和还原。利用olex2软件,选择shelxt程序以直接法解析结构,选择shelxl程序以全矩阵最小二乘法对结果进行精修。
[0078]
本实施例所制备的灰黄霉素i晶型单晶的晶体结构数据如表1所示,其晶胞堆积图如图4(1)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图3(1)所示。
[0079]
表1.灰黄霉素i、ii和iii晶型的晶体结构参数
[0080][0081]
实施例2
[0082]
本实施例提供一种灰黄霉素ii晶型单晶的制备方法,步骤如下:
[0083]
步骤1)将少许灰黄霉素粉末放置于玻片上,再置于样品放置台,将linkam热台升温到230℃,将灰黄霉素粉末加热10s以熔融,得到灰黄霉素熔体,然后快速转移到温度预设为110℃的热台,放置48h,得到灰黄霉素iii晶型的多晶样品(形貌为球晶);再将该样品转移到温度预设为110℃的linkam热台上,以100℃/min升温速率快速升温到205℃(该温度为灰黄霉素iii晶型的熔点),得到ii晶型和iii晶型的混合物。
[0084]
步骤2)将该混合物以10℃/min升温至214℃,熔融大部分样品;再以1℃/min升温缓慢升温至215℃附近(灰黄霉素ii晶型的熔点)。
[0085]
步骤3)若观察到剩余灰黄霉素ii晶型的1-10颗单晶晶核,则降温到205℃,恒温,
进行单晶培养,得灰黄霉素ii晶型单晶。
[0086]
步骤4)若所有晶体完全熔融,仍没有获得1-10颗灰黄霉素i晶型的单晶晶核,则重复步骤1)和2),升温到214℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得灰黄霉素ii晶型的1-10颗单晶晶核,执行步骤3)。
[0087]
本实施例所制备的灰黄霉素ii晶型单晶的显微照片如图2(2)所示。
[0088]
对灰黄霉素ii晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图3(2)所示。
[0089]
对所制备的灰黄霉素ii晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的灰黄霉素ii晶型单晶的晶体结构数据如表1所示,其晶胞堆积图如图4(2)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图3(2)所示。
[0090]
采用步骤1)中的灰黄霉素ii晶型多晶样品制备方法,是因为灰黄霉素ii晶型难以在熔体中自发成核,因此先制备灰黄霉素iii晶型,利用其在170-190℃发生的固-固晶型转变获得灰黄霉素ii晶型。
[0091]
此外,灰黄霉素ii晶型还可以通过引入晶种的方法,在过冷熔体中引入灰黄霉素ii晶型的晶种,再进行上述精细控温直至剩余1-10颗单晶晶核,然后降温到合适的温度进行培养,获得单晶。
[0092]
实施例3
[0093]
本实施例提供一种灰黄霉素iii晶型单晶的制备方法,步骤如下:
[0094]
步骤1)将少许灰黄霉素粉末状样品放置于玻片上,再置于样品放置台,将linkam热台升温到230℃,将灰黄霉素粉末状样品加热10s、完全熔融,然后快速转移到温度预设为110℃的另一个热台上,放置48h,获得灰黄霉素iii晶型的多晶样品(球晶),作为晶种。
[0095]
步骤2)另取少许灰黄霉素粉末状样品放置于玻片上,再置于样品放置台,将linkam从30℃热台升温到230℃,将灰黄霉素粉末状样品加热10s、完全熔融,降温至200℃,得过冷熔体,然后用针引入灰黄霉素iii晶型的晶种,获得多晶样品。
[0096]
步骤3)将多晶样品以10℃/min升温至204℃。再以1℃/min升温速率,将样品温度缓慢升到206℃附近(灰黄霉素iii的熔点)。
[0097]
步骤4)若观察到剩余1-10颗灰黄霉素iii的单晶晶核,则降温至200℃,恒温培养,得灰黄霉素iii晶型单晶。
[0098]
步骤5)若所有晶体完全熔融,仍没有获得1-10颗灰黄霉素i晶型的单晶晶核,则重复步骤2)和3),升温到204℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1-10颗灰黄霉素iii的单晶晶核,执行步骤4)。
[0099]
本实施例所制备的灰黄霉素iii晶型单晶的显微照片如图2(3)所示。
[0100]
对灰黄霉素iii晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图3(3)所示。
[0101]
对所制备的灰黄霉素iii晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的灰黄霉素iii晶型单晶的晶体结构数据如表1所示,其晶胞堆积图如图4(3)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图3(3)所示。
[0102]
本实施例步骤1)中,在200℃向灰黄霉素过冷熔体中引入iii晶型晶种的原因如下:
[0103]
a.在更低的温度(如170-190℃),将灰黄霉素iii晶型的晶种引入过冷熔体后,灰黄霉素iii晶型可能转变为灰黄霉素ii晶型;
[0104]
b.向过冷熔体中引入灰黄霉素iii晶型晶种的温度,可以设置为200℃以上、iii晶型熔点之下的其他温度;
[0105]
c.高于200℃时,灰黄霉素的过冷熔体自发成核困难,所以其他晶型很难自发成核。
[0106]
维罗非尼的分子结构如下所示:
[0107][0108]
实施例4
[0109]
本实施例提供一种维罗非尼β晶型单晶的制备方法,步骤如下:
[0110]
步骤1)将少许维罗非尼粉末放置于玻片上,再置于温度预设为277℃的linkam热台上,恒温1s以完全熔融样品后,转移至170℃恒温,获得维罗非尼α、β、γ的多晶共生样品(各晶型形貌均为球晶),如图5所示。
[0111]
步骤2)另取少许维罗非尼粉末放置于玻片上,再置于温度预设为277℃的linkam热台上,恒温1s以完全熔融样品,然后以100℃/min降温到245℃(本实施例的t
1
),得过冷熔体,引入维罗非尼β晶型晶种,获得β晶型多晶样品。
[0112]
步骤3)将多晶样品以10℃/min升温至255℃,熔融大部分样品;再以1℃/min升温至255.4℃附近(维罗非尼的β晶型熔点是255℃)。
[0113]
步骤4)若观察到剩余1颗维罗非尼β晶型的单晶晶核,以100℃/min降温至252℃恒温培养,获得维罗非尼β型单晶。
[0114]
步骤5)若多晶样品完全熔融,仍未获得1颗单晶晶核,则重复步骤2)和3),升温至255℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗维罗非尼β晶型的单晶晶核,执行步骤4)。
[0115]
本实施例所制备的维罗非尼β晶型的单晶的显微照片如图6(1)所示。
[0116]
对维罗非尼β晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图7(1)所示。
[0117]
对所制备的维罗非尼β晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的维罗非尼β晶型单晶的晶体结构数据如表2所示,其晶胞堆积图如图8(1)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图7(1)所示。
[0118]
表2.维罗非尼β和γ晶型的晶体结构参数
[0119][0120]
本实施例中,采用引入晶种法的原因是,在维罗非尼β自发成核的温度,容易同时产生维罗非尼α单晶晶核而无法获得纯的维罗非尼β晶型,故不采用自发成核进行后续操作。
[0121]
实施例5
[0122]
本实施例提供一种维罗非尼γ型单晶的制备方法,步骤如下:
[0123]
步骤1)将少许维罗非尼粉末放置于玻片上,再置于温度预设为277℃的linkam热台上,恒温1s以完全熔融样品,然后以100℃/min降温至170℃恒温,获得维罗非尼α、β、γ的多晶共生样品(各晶型形貌均为球晶),如图5所示。
[0124]
步骤2)另取少许维罗非尼粉末放置于玻片上,再置于温度预设为277℃的linkam热台上,恒温1s以完全熔融样品,然后以100℃/min降温到215℃,得过冷熔体,然后引入维罗非尼γ晶型晶种,获得γ晶型多晶样品。
[0125]
步骤3)将多晶样品以10℃/min升温至223℃;再以1℃/min缓慢升温到224℃(维罗非尼的γ晶型熔点是224℃)附近。
[0126]
步骤4),若观察到剩余1颗维罗非尼γ晶型的单晶晶核,则降温至215℃,恒温培养,获得维罗非尼γ晶型单晶。
[0127]
步骤5)若多晶样品完全熔融,仍未获得1颗γ晶型单晶晶核,则重复步骤2)和3),升温至223℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗γ晶型单晶晶核,执行步骤4)。
[0128]
本实施例所制备的维罗非尼γ晶型的单晶的显微照片如图6(2)所示。
[0129]
对维罗非尼γ晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图7(2)所示。
[0130]
对所制备的维罗非尼γ晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的维罗非尼γ晶型单晶的晶体结构数据如表2所示,其晶胞堆积图如图8(2)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图7(2)所示。
[0131]
硝苯地平的分子结构如下所示:
[0132][0133]
实施例6
[0134]
本实施例提供一种硝苯地平α晶型单晶的制备方法,步骤如下:
[0135]
步骤1)将少许硝苯地平粉末放置于玻片上,再置于linkam热台上。
[0136]
步骤2)从30℃以100℃/min升温至172℃;再以10℃/min升温至175℃附近,再以1℃/min升温至176.4℃附近(硝苯地平α晶型的熔点)。
[0137]
步骤3)若观察到剩余1颗α晶型单晶晶核,则以150℃/min迅速降温至170℃恒温培养α晶型单晶。
[0138]
步骤4)若粉末样品完全熔融,仍未获得1颗α晶型单晶晶核,则重复步骤1)和2),升温至172℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗α晶型单晶晶核,执行步骤3)。
[0139]
本实施例所制备的硝苯地平α晶型单晶的显微照片如图9(1)所示。
[0140]
对硝苯地平α晶型粉末样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图10所示。
[0141]
对所制备的硝苯地平α晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的硝苯地平α晶型单晶的晶体结构数据如表3所示,其晶胞堆积图如图11(1)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图10所示。
[0142]
表3.硝苯地平α、β和γ晶型的晶体结构参数
[0143]
[0144]
实施例7
[0145]
本实施例提供一种硝苯地平β晶型单晶的制备方法,步骤如下:
[0146]
步骤1)将少许硝苯地平粉末放置于玻片上,再置于温度预设为180℃的linkam热台上,恒温10s以完全熔融样品,然后降温到100℃恒温,获得硝苯地平β晶型和γ晶型的多晶共生样品,作为晶种。
[0147]
步骤2)另取少许硝苯地平粉末放置于玻片上,再置于温度预设为180℃的linkam热台上,恒温10s以完全熔融样品,然后以100℃/min降温至160℃,得过冷熔体,引入硝苯地平β晶型晶种,获得β晶型多晶样品。
[0148]
步骤3)将多晶样品以10℃/min升温至164℃。再以1℃/min缓慢升到165℃附近(硝苯地平β晶型的熔点是165℃),熔融大部分晶体。
[0149]
步骤4)若观察到剩余1颗硝苯地平β晶型的单晶晶核,降温至155-160℃,恒温培养,得硝苯地平β晶型单晶。
[0150]
步骤5)若多晶样品完全熔融,仍未观察到硝苯地平β晶型单一单晶晶核剩余,则重复步骤2)和3),升温至164℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗硝苯地平β晶型的单晶晶核,执行步骤4)。
[0151]
本实施例所制备的硝苯地平β晶型单晶的显微照片如图9(2)所示。
[0152]
对所制备的硝苯地平β晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的硝苯地平β晶型单晶的晶体结构数据如表3所示,其晶胞堆积图如图11(2)所示。
[0153]
实施例8
[0154]
本实施例提供一种硝苯地平γ晶型单晶的制备方法,步骤如下:
[0155]
步骤1)将少许硝苯地平粉末放置于玻片上,再置于温度预设为180℃的linkam热台上,恒温10s以完全熔融样品,然后降温到100℃恒温,获得硝苯地平β晶型和γ晶型的多晶共生样品,作为晶种。
[0156]
步骤2)另取少许硝苯地平粉末放置于玻片上,再置于温度预设为180℃的linkam热台上,恒温10s以完全熔融样品,然后以100℃/min降温至120℃,得过冷熔体,引入硝苯地平γ晶型晶种,获得γ晶型多晶样品。
[0157]
步骤3)将多晶样品以10℃/min升温至137℃。再以1℃/min缓慢升到138℃附近(硝苯地平γ晶型的熔点是138℃),熔融大部分晶体。
[0158]
步骤4)若观察到剩余1颗硝苯地平γ晶型的单晶晶核,降温至125℃,恒温培养,得硝苯地平γ晶型单晶。
[0159]
步骤5)若多晶样品完全熔融,仍未观察到硝苯地平γ晶型单一单晶晶核剩余,则重复步骤2)和3),升温至137℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗硝苯地平γ晶型的单晶晶核,执行步骤4)。
[0160]
本实施例所制备的硝苯地平γ晶型单晶的显微照片如图9(3)所示。
[0161]
对所制备的硝苯地平γ晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的硝苯地平γ晶型单晶的晶体结构数据如表3所示,其晶胞堆积图如图11(3)所示。
[0162]
本实施例中,硝苯地平γ晶型在100℃恒温时,容易形成针状晶体,但由于此时无
法除去样品中的硝苯地平β晶(熔点高于硝苯地平γ晶),故需使用引入晶种的方法获得纯的硝苯地平γ晶型。
[0163]
烟酰胺的分子结构如下所示:
[0164][0165]
实施例9
[0166]
本实施例提供一种烟酰胺α晶型单晶的制备方法,步骤如下:
[0167]
步骤1)将少许烟酰胺粉末放置于玻片上,再置于linkam热台上。
[0168]
步骤2)从30℃以100℃/min的升温速率升温至126℃;再以10℃/min的升温速率升温至128℃附近;再以1℃/min的升温速率升温至129℃附近(烟酰胺α晶型的熔点为129℃)。
[0169]
步骤3)若观察到剩余1颗烟酰胺α晶型单晶晶核,则以150℃/min的降温速率迅速降温至127℃恒温培养α晶型单晶。
[0170]
步骤4)若粉末样品完全熔融,仍未获得单一烟酰胺α晶型单晶晶核,则重复步骤1)和2),升温至128℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗烟酰胺α晶型单晶晶核,执行步骤3)。
[0171]
本实施例所制备的烟酰胺α晶型单晶的显微照片如图12(1)所示。
[0172]
对烟酰胺α晶型粉末样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图13(1)所示。
[0173]
对所制备的烟酰胺α晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的烟酰胺α晶型单晶的晶体结构数据如表4所示,其晶胞堆积图如图14(1)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图13(1)所示。
[0174]
表4.烟酰胺α、β、γ、δ、ε和ζ晶型的晶体结构数据
[0175][0176][0177]
实施例10
[0178]
本实施例提供一种烟酰胺β型单晶的制备方法,步骤如下:
[0179]
步骤1)将少许烟酰胺粉末放置于玻片上,再置于预设温度为140℃的热台上恒温10s完全熔融样品,然后将样品转移到预设温度为60℃的linkam热台上,恒温至生成烟酰胺ζ晶型。继续放置,直至观察到烟酰胺ζ晶型表面转变为烟酰胺β晶型。
[0180]
步骤2)将上述烟酰胺β晶型多晶样品以100℃/min的升温速率升温至114℃,熔融可能出现的烟酰胺γ、δ和ζ晶型。随后,以10℃/min的升温速率升温至115℃附近,再以1℃/min的升温速率升温至116℃附近(烟酰胺β晶型的熔点为116.5℃)。
[0181]
步骤3)若观察到剩余1颗烟酰胺β晶型的单晶晶核,则以150℃/min的降温速率降温至110℃,恒温培养,得烟酰胺β晶型单晶。
[0182]
步骤4)若β晶型多晶样品完全熔融,仍未获得单一单晶晶核,则重复步骤1)和2),升温至115℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗烟酰胺β晶型的单晶晶核,执行步骤3)。
[0183]
本实施例所制备的烟酰胺β晶型单晶的显微照片如图12(2)所示。
[0184]
对烟酰胺β晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图13(2)所示。
[0185]
对所制备的烟酰胺β晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的烟酰胺β晶型单晶的晶体结构数据如表4所示,其晶胞堆积图如图14(2)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图13(2)所示。
[0186]
实施例11
[0187]
一种烟酰胺γ晶型单晶的制备方法,步骤如下:
[0188]
步骤1)将少许烟酰胺粉末放置于玻片上,再置于预设温度为140℃的热台上恒温10s完全熔融样品,然后将样品转移到预设温度为70℃的linkam热台上,恒温至生成烟酰胺γ晶型。
[0189]
步骤2)将多晶样品以100℃/min升温至111℃,再以10℃/min的升温速率升温至113℃附近,再以0.5℃/min的升温速率升温至114℃附近(烟酰胺γ晶型的熔点为114℃)。
[0190]
步骤3)若观察到剩余1颗烟酰胺γ晶型的单晶晶核,则以150℃/min的降温速率降温至110℃,恒温培养,得烟酰胺γ晶型单晶。
[0191]
步骤4)若γ晶型多晶样品完全熔融,仍未获得单一单晶晶核,则重复步骤1)和2),升温至113℃后,在0.1-0.5℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗烟酰胺γ晶型的单晶晶核,执行步骤3)。
[0192]
本实施例中,若在温度到达114℃之前,所有晶体都已熔融,说明该晶体不是烟酰胺γ晶型,则重复步骤1)和2),若晶体中出现的最高熔点介于114-116℃之间,则可用于烟酰胺γ晶型单晶制备。
[0193]
本实施例所制备的烟酰胺γ晶型单晶的显微照片如图12(3)所示。
[0194]
对烟酰胺γ晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图13(3)所示。
[0195]
对所制备的烟酰胺γ晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的烟酰胺γ晶型单晶的晶体结构数据如表4所示,其晶胞堆积图如图14(3)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图13(3)所示。
[0196]
实施例12
[0197]
本实施例提供一种烟酰胺δ晶型单晶的制备方法,步骤如下:
[0198]
步骤1)将少许烟酰胺粉末放置于玻片上,再置于预设温度为140℃的热台上恒温10s完全熔融样品,然后将样品转移到预设温度为70℃的linkam热台上,恒温放置,有较大概率会获得烟酰胺δ晶型。
[0199]
步骤2)将多晶样品以100℃/min升温至107℃,再以10℃/min的升温速率升温至109℃附近,再以0.1℃/min的升温速率升温至110℃附近(烟酰胺δ晶型的熔点为110℃)。
[0200]
步骤3)若观察到剩余1颗烟酰胺δ晶型的单晶晶核,则以150℃/min的降温速率降温至108℃,恒温培养,获得烟酰胺δ晶型单晶。
[0201]
步骤4)若δ晶型多晶样品完全熔融,仍未获得单一单晶晶核,则重复步骤1)和2),升温至109℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗烟酰胺δ晶型的单晶晶核,执行步骤3)。
[0202]
本实施例中,若晶体中出现的最高熔点介于110-113℃之间,则可用于烟酰胺δ晶型单晶制备。
[0203]
本实施例所制备的烟酰胺δ晶型单晶的显微照片如图12(4)所示。
[0204]
对烟酰胺δ晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图13(4)所示。
[0205]
对所制备的烟酰胺δ晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的烟酰胺δ晶型单晶的晶体结构数据如表4所示,其晶胞堆积图如图14(4)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图13(4)所示。
[0206]
实施例13
[0207]
本实施例提供一种烟酰胺ε晶型单晶的制备方法,步骤如下:
[0208]
步骤1)将少许烟酰胺粉末放置于玻片上,再置于预设温度为140℃的热台上恒温10s完全熔融样品,然后将样品转移到预设温度为70℃的linkam热台上,恒温放置,获得烟酰胺ε晶型。
[0209]
步骤2)将多晶样品以100℃/min升温至106℃,再以10℃/min的升温速率升温至107℃附近,再以0.1℃/min的升温速率升温至108℃附近(烟酰胺ε晶型的熔点为108℃)。
[0210]
步骤3)若观察到剩余1颗烟酰胺ε晶型的单晶晶核,则以150℃/min的降温速率降温至106℃,恒温培养,获得烟酰胺ε晶型单晶。
[0211]
步骤4)若ε晶型多晶样品完全熔融,仍未获得单一单晶晶核,则重复步骤1)和2),升温至107℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗烟酰胺ε晶型的单晶晶核,执行步骤3)。
[0212]
本实施例所制备的烟酰胺ε晶型单晶的显微照片如图12(5)所示。
[0213]
对烟酰胺ε晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图13(5)所示。
[0214]
对所制备的烟酰胺ε晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的烟酰胺ε晶型单晶的晶体结构数据如表4所示,其晶胞堆积图如图14(5)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图13(5)所示。
[0215]
本实施例中,若晶体中出现的最高熔点介于108-110℃之间,则可用于烟酰胺ε晶
型单晶制备。
[0216]
该晶型在玻璃基质上出现的概率低于1%,在铝制基质上出现概率较大。
[0217]
实施例14
[0218]
本实施例提供一种烟酰胺ζ晶型单晶的制备方法,步骤如下:
[0219]
步骤1)将少许烟酰胺粉末放置于玻片上,再置于预设温度为140℃的热台上恒温10s完全熔融样品,然后将样品转移到预设温度为70℃的linkam热台上,一旦观察到烟酰胺ζ晶型晶核,立即以150℃/min迅速升温至100℃,越过60-80℃温度区间(该区间烟酰胺ζ晶型非常容易转变为β晶型)。
[0220]
步骤2)再以100℃/min升温速率升温至102℃,再以10℃/min的升温速率升温至103℃附近,随后以0.1℃/min升温速率升温至104℃附近(烟酰胺ε晶型的熔点为104℃)。
[0221]
步骤3)若观察到剩余1颗烟酰胺ζ晶型的单晶晶核,则以150℃/min的降温速率降温至102℃,恒温培养,获得烟酰胺ζ晶型单晶。
[0222]
步骤4)若ζ晶型多晶样品完全熔融,仍未获得单一单晶晶核,则重复步骤1)和2),升温至103℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗烟酰胺ζ晶型的单晶晶核,执行步骤3)。
[0223]
本实施例所制备的烟酰胺ζ晶型单晶的显微照片如图12(6)所示。
[0224]
对烟酰胺ζ晶型多晶样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图13(6)所示。
[0225]
对所制备的烟酰胺ζ晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的烟酰胺ζ晶型单晶的晶体结构数据如表4所示,其晶胞堆积图如图14(6)所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图13(6)所示。
[0226]
r-克唑替尼的分子结构如下所示:
[0227][0228]
实施例15
[0229]
本实施例提供一种r-克唑替尼α晶型单晶的制备方法,步骤如下:
[0230]
步骤1)将少许r-克唑替尼粉末放置于玻片上,再置于linkam热台上。
[0231]
步骤2)从30℃以100℃/min升温至200℃,再以10℃/min的升温速率升温至204℃附近,再以1℃/min升温至204.4℃附近(r-克唑替尼α晶型的熔点为205℃)。
[0232]
步骤3)若观察到中剩余1颗r-克唑替尼α晶型单晶晶核,则以150℃/min迅速降温
至180℃恒温培养α晶型单晶。
[0233]
步骤4)若粉末样品完全熔融,仍未获得1颗α晶型单晶晶核,则重复步骤1)和2),升温至204℃后,在0.1-1℃/min的升温速率内,根据晶核的变化调整升温速率,直至获得1颗r-克唑替尼α晶型单晶晶核,执行步骤3)。
[0234]
本实施例所制备的r-克唑替尼α晶型单晶的显微照片如图15所示。
[0235]
对r-克唑替尼α晶型的粉末样品进行粉末x射线衍射测定,测试条件与实施例1相同,测试谱图如图16所示。
[0236]
对所制备的r-克唑替尼α晶型单晶进行x射线单晶衍射测定,测试条件、数据处理方法与实施例1相同,所制备的r-克唑替尼α晶型单晶的晶体结构数据如表5所示,其晶胞堆积图如图17所示,根据其单晶结构数据拟合得到的粉末x射线衍射谱图如图16所示。
[0237]
表5.r-克唑替尼α晶型的晶体结构参数
[0238][0239]
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
[0240]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签: 烟酰胺

 热门咨询
热门咨询




tips