
 商标分类
商标分类  商标转让
商标转让 
一种石墨烯增韧二维高氮材料掺杂硝胺氧化剂及制备方法与流程
 2021-01-30 17:01:56|
2021-01-30 17:01:56| 315|
315| 起点商标网
起点商标网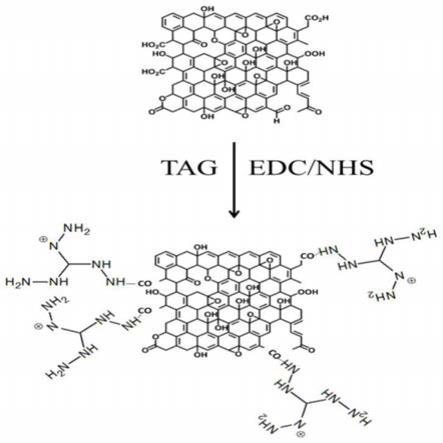
[0001]
本发明属于含能材料技术领域,涉及一种石墨烯增韧二维高氮材料掺杂硝胺氧化剂及制备方法,具体涉及一种石墨烯增韧二维高氮材料掺杂硝胺氧化剂(rdx、hmx、bchmx、cl-20)的制备方法。
背景技术:
[0002]
近年来,硝铵类含能材料(如rdx、hmx、bchmx、cl-20)因具有高的能量密度而受到广泛关注,但由于存在多晶型转变问题,其在极性溶剂或增塑剂存在情况下的使用仍有较大限制(yan ql,zeman s,elbeih a,song zw,m
á
lek j.the effect of crystal structure on the thermal reactivity of cl-20and its c4 bonded explosives(i):thermodynamic properties and decomposition kinetics.j therm anal calorim 2013;112(2):823-836)。以六硝基六氮杂异伍兹烷(cl-20)为例,这是一种多晶型炸药,常温常压下存在α-、β-、γ-、及ε-四种晶型。,为了克服硝铵类含能材料转晶的问题,研究人员采用了各种策略来降低硝铵类含能材料的感度,提高其热稳定性。研究表明,掺杂碳纳米材料可以优化ε-cl-20晶体的表面形态和粒径分布,从而降低其感度(rossi c.two decades of research on nano-energetic materials.propellants,explosion,pyrotechnics 2014;39(3):323-327.)。碳纳米材料是最典型的一种,其优异的物理性能,如高的比表面积,导电率和导热率等,其常温下的导热率高达5000w/m
·
k,理想的单层石墨烯结构比表面积达到2630m2/g,有助于降低含能晶体的感度。其中,go包含大量的含氧官能团,被认为是具有一定能级能量的掺杂材料(li zm,wang y,zhang yq.cl-20hosted in graphene foam as a high energy material with low sensitivity.rsc adv 2015;5(120):98925
–
98928),因此在高能材料(ems)领域引起了广泛的关注。go的另一个特点是它可以在含能材料分解前稳定其晶型,并在ems的分解和燃烧过程中催化液相或气相反应(he w,guo jh,cao ck,liu xk,lv jy,chen sw,liu pj,yan ql.catalytic reactivity of graphene oxide stabilized transition metal complexes of triaminoguanidine on thermolysis of rdx.j phys chem c 2018;122(26):14714-14724)。
[0003]
目前,各种硝铵类高能晶体主要通过与go物理混合的方法进行改性(yan ql,gozin m,zhao fq,cohen a,pang sp.high energetic compositions based on functionalized carbon nanomaterials.nanoscale 2016;8(9):4799-4851)。结果表明,go不会改变ems的晶体结构,而只会改变其表面形态,且其在安全性和能量释放率方面具有比其他碳纳米材料(例如富勒烯和碳纳米管)具有更好的协同增强作用(wang s,an cw,wang j.reduce the sensitivity of cl-20by improving thermal conductivity through carbon nanomaterials.nanoscale 2018;13(1):85)。研究表明,cl-20晶体可以使用水悬浮法同时被氧化石墨烯(rgo)和碳纳米管(cnt)以一定的比例包覆,并且rgo/cnt的加入可以提高cl-20晶体的热导率,从而降低外界刺激下形成热点的可能性。通过这种方式,在不改变能量密度和晶体结构的情况下,撞击感度被显著降低(h
50
从17.3厘米增加到
68.7厘米)。
[0004]
在高能晶体表面上物理包覆石墨烯可以显著改善其稳定性,但高能量密度一直是储能或含能材料(ems)发展的关键目标。除了探索具有高化学能的新型ems外,传统ems在更高密度下的组装也是值得期待的。已有研究表明,tagp层的叠加会产生分子水平压缩效应,从而导致具有改变构象ems分子密度更高。在溶剂中压缩形成的含能晶体的单位细胞参数非常接近于在0.2gpa的压力下观察到结果。这表明含能分子被困在tagp层,从而得到更高的密度(例如qy-hmx达到2.13g/cm3)和更好的热稳定性(yan ql,yang zj,zhang xx,lyu jy,he w,huang s,liu pj,zhang cy,zhang qh,he gq,nie fd.high density assembly of energetic molecules under the constraint of defected 2d materials.j.mater.chem.a 2019;7:17806-17814)。部分比例受约束的qy-hmx晶体没有缺陷,且在加热时没有观察到多态转变和熔点。实验和相关的计算表明,最好的改性hmx晶体的爆炸速度达到10.40km/s,爆压达到53.9gpa。比冲达到约292秒,这些提高的性能使其成为未来空间探索中很有前途的推进剂组件。
[0005]
综上,掺杂tagp可用于增强分子水平压缩效应,从而提高硝铵类含能晶体的密度,go的引入也可在稳定含能晶体晶型的同时催化硝铵晶体的热分解。通过优化,go-tagp的掺杂可以进一步实现硝铵类含能晶体的优良热稳定性及高密度。
技术实现要素:
[0006]
要解决的技术问题
[0007]
为了避免现有技术的不足之处,本发明提出一种石墨烯增韧二维高氮材料掺杂硝胺氧化剂及制备方法,以解决硝铵类含能晶体热稳定性较差和现有技术改性降低含能晶体能量密度问题。
[0008]
技术方案
[0009]
一种石墨烯增韧二维高氮材料掺杂硝胺氧化剂,其特征在于:石墨烯增韧二维高氮材料的质量含量为0.5wt%~1.5wt%,硝胺氧化剂的质量含量为98.5wt%~99.5wt%;所述石墨烯增韧二维高氮材料为:氧化石墨烯与三氨基胍硝酸盐和40%乙二醛溶液形成的对硝胺氧化剂具有限域作用的二维结构。
[0010]
所述硝胺氧化剂包括但不限于:rdx、hmx、bchmx或cl-20高能炸药。
[0011]
一种制备所述石墨烯增韧二维高氮材料掺杂硝胺氧化剂的方法,其特征在于步骤如下:
[0012]
步骤1:将含氧量≥45%的氧化石墨烯go分散到有机溶剂中,超声处理60min,得到氧化石墨烯悬浮液;在温度为70~75℃条件下,将1-乙基-3-(3-二甲氨丙基)碳二亚胺盐酸盐edc和n-羟基丁二酰亚胺nhs的去离子水混合液加入到go的分散液中,在此温度下保持30min,得到活化羧基的go分散液;在温度为70~75℃条件下,将三氨基胍硝酸盐tagn加入到上述混合液中,反应4h,得到go-tag黑色絮状沉淀,自然降温到室温后抽滤并干燥;
[0013]
所述预制edc和nhs的去离子水混合液中edc和nhs的质量比为4:3;
[0014]
步骤2:70~75℃时,将go-tag分散在二甲基亚砜dmso中,得到go-tag的dmso分散液;
[0015]
步骤3:再加入与cl-20摩尔比为1:8~1:3所对应质量的tagn,待充分溶解后,得到
含有tagn、go-tag的悬浮液;
[0016]
步骤4:再将ε-cl-20粉末加入到3所得悬浮液中,待cl-20完全溶解后,缓慢滴加与tagn摩尔比为1:1~1:1.5对应体积的40%乙二醛溶液,反应60~70min;
[0017]
步骤5:将与dmso体积比为1:1的去离子水缓慢滴加到s4混合液中,保温10~15分钟后,40~45℃蒸馏水冲洗4遍;抽滤和真空干燥后,得到以氧化石墨烯-三氨基胍硝酸盐复合物go-tagp掺杂的cl-20含能材料;
[0018]
其中,go-tag的掺杂量为0.5~1.5wt%;步骤1中go与edc的质量比为5:1,与nhs质量比为20:3,加入tagn的质量与最初加入go质量比为10:1。
[0019]
所述有机溶剂为去离子水。
[0020]
所述dmso相对密度为1.098-1.102g/cm3。
[0021]
有益效果
[0022]
本发明提出的一种石墨烯增韧二维高氮材料掺杂硝胺氧化剂及制备方法,通过引入氧化石墨烯,降低了操作过程中的风险;制备出稳定性、密度较高的硝铵类含能材料,即go-tagp掺杂硝铵类含能材料,其晶体密度提高、分子结构未发生改变;转晶温度的提高(或转晶过程的消失)表明其热稳定性得到提高;
[0023]
当以go作为掺杂材料时,以溶剂-非溶剂法得到的共晶具有层状结构,可以获得更高的晶体密度,其次,利用go高的导热率和大的比表面积的特点,在提高硝铵类晶体的热稳定性方面具有突出贡献;同时,内嵌的go薄膜又能提高晶体的刚度,并且go对含能氧化剂的分解有一定的催化效应,不但降低了热点起爆的几率,使得最终复合含能材料的机械感度大幅降低,还可以在提高热稳定性的同时提高分解效率。当以聚合的tagp作为约束材料时,含能分子可被困在tagp层,从而得到更高密度的改性含能晶体,并且具有更好的热稳定性。
[0024]
通过控制go的含量(0.5~1.5wt%)来调控复合晶体结构,使其热稳定性得到优化。上述优化后工艺所制备的钝感含能材料的密度得到提高,且热分析结果表明其热稳定性较高。本发明得到的钝感含能材料的制备工艺简单、热稳定性高、晶体密度较高,主要是因为该方法制备的含能材料具有较高的热导率和催化效率、转晶温度提高、热稳定性得到一定程度的提高,对将来硝铵类含能材料的广泛应用打下一定基础。
附图说明
[0025]
图1为本发明实施例1、2、3、4、5、6、7、8和9中go-tag结合方式结构模型机理;
[0026]
图2为本发明实施例2go-tagp掺杂ε-cl-20含能材料的扫描电子显微镜和透射电子显微镜图;
[0027]
图3为本发明实施例4go-tagp掺杂ε-cl-20含能材料的扫描电子显微镜图;
[0028]
图4为本发明实施例5go-tagp掺杂ε-cl-20含能材料的扫描电子显微镜和透射电子显微镜图;
[0029]
图5为本发明实施例6go-tagp掺杂ε-cl-20含能材料的扫描电子显微镜;
[0030]
图6为本发明实施例7go-tagp掺杂ε-cl-20含能材料的扫描电子显微镜图;
[0031]
图7为本发明实施例8go-tagp掺杂ε-cl-20含能材料的扫描电子显微镜和透射电子显微镜图;
[0032]
图8为本发明实施例9go-tagp掺杂β-hmx含能材料的扫描电子显微镜图;
[0033]
图9为本发明实施例2、5、8和9制备的掺杂ε-cl-20/hmx含能材料在不同条件下的dsc/tg曲线;
[0034]
图10为本发明实施例2、5、8和9制备的掺杂ε-cl-20/hmx含能材料在不同掺杂物下xrd曲线。
具体实施方式
[0035]
现结合实施例、附图对本发明作进一步描述:
[0036]
石墨烯增韧二维高氮材料掺杂硝胺氧化剂的制备方法,硝胺氧化剂特指rdx、hmx、bchmx和cl-20等高能炸药;在掺杂过程中,石墨烯增韧二维高氮材料(氧化石墨烯与三氨基胍硝酸盐和40%乙二醛溶液形成的对硝胺氧化剂具有限域作用的二维结构)的质量含量为0.5wt%~1.5wt%,硝胺氧化剂的质量含量为98.5wt%~99.5wt%;go具有优异的导电率,可以优化硝胺晶体的表面形态和粒径分布,从而降低其感度;二维三氨基胍-乙二醛聚合物(tagp)层的叠加会产生分子水平压缩效应,从而导致具有改变构象的硝胺氧化剂密度更高。此制备方法特征在于所述制备方法包括以下步骤:
[0037]
s1.将含氧量≥45%的氧化石墨烯(go)分散到去离子水(或其他有机溶剂)中,超声处理60min,得到氧化石墨烯悬浮液;在温度为70~75℃条件下,将1-乙基-3-(3-二甲氨丙基)碳二亚胺盐酸盐(edc)和n-羟基丁二酰亚胺(nhs)的去离子水混合液加入到go的分散液中,在此温度下保持30min,得到活化羧基的go分散液;在温度为70~75℃条件下,将三氨基胍硝酸盐(tagn)加入到上述混合液中,反应4h,得到go-tag黑色絮状沉淀,自然降温到室温后抽滤并干燥;
[0038]
所述预制edc和nhs的去离子水混合液中edc和nhs的质量比为4:3;
[0039]
在70~75℃下,将edc和nhs的一定量去离子水混合液加入go分散液中,在此温度下反应30min可得到活化羧基的go分散液,使go片层结构侧面的羧基变得活泼,易与tagn上的氨基发生脱水缩合,形成较强的肽键(-co-nh-),以此生成go作为端基的交联体;
[0040]
其中,c2h2o2的质量分数为40%,在计算过程中以100%的质量分数计算,滴加时计算所需40%c2h2o2量过少,无法精确量取,因此按一滴0.05ml计算需加的试剂量。
[0041]
s2.70~75℃时,将一定量s1所制备go-tag分散在二甲基亚砜(dmso)中,得到go-tag的dmso分散液;
[0042]
s3.在s2得到的溶液中加入与cl-20摩尔比为1:8~1:3所对应质量的tagn,待充分溶解后,得到含有tagn、go-tag的悬浮液;
[0043]
s4.再将一定质量的ε-cl-20粉末加入到3所得悬浮液中,待cl-20完全溶解后,缓慢滴加与tagn摩尔比为1:1~1:1.5对应体积的40%乙二醛溶液,反应60~70min;
[0044]
s5.将与dmso体积比为1:1的去离子水缓慢滴加到s4混合液中,保温10~15分钟后,40~45℃蒸馏水冲洗4遍;抽滤和真空干燥后,得到以氧化石墨烯-三氨基胍硝酸盐复合物(go-tagp)掺杂的cl-20含能材料;
[0045]
其中,go-tag的理论掺杂量为0.5wt%,1wt%,1.5wt%;步骤s1中go与edc的质量比为5:1,与nhs质量比为20:3,加入tagn的质量与最初加入go质量比为10:1;
[0046]
滴加反溶剂时,蠕动泵的转速为150r/min,加入去离子水体积与dmso为1:1时基本可以使产品完全析出;
[0047]
由于dmso熔点较低(18~20℃),抽滤和干燥过程中洗涤用40℃去离子水。
[0048]
溶解硝铵类含能晶体所选溶剂为dmso;
[0049]
所选反溶剂为去离子水;
[0050]
所述dmso相对密度为1.098-1.102g/cm3。
[0051]
实施例1
[0052]
一种石墨烯增韧二维高氮材料掺杂ε-cl-20晶体的制备方法,包括如下工艺步骤:
[0053]
氧化石墨烯悬浮液的配制:
[0054]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0055]
配制edc与nhs的去离子水溶液:
[0056]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0057]
go-tag的制备:
[0058]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0059]
以go-tag的掺杂量为0.5wt%,cl-20与go-tag的摩尔比为3:1,所加入tagn与乙二醛溶液摩尔比为1:1进行掺杂,先将11.6mg go-tag分散于10ml dmso中(在所需产量不同的情况下,dmso的加入量为刚好使cl-20溶解为宜),体系温度保持在70~75℃,在磁力搅拌下得到go-tag的dmso分散液;
[0060]
再向上述混合液中加入250mg的tagn,磁力搅拌10min使混合更加均匀,再加入1972.5mgε-cl-20粉末,待加入的cl-20完全溶解后,缓慢滴加3滴40%乙二醛溶液,反应60min;
[0061]
将约10ml去离子水缓慢滴加到上述混合液中(蠕动泵的转速为150r/min),边滴加边搅拌,反应温度保持在70~75℃,得到以go-tagp掺杂ε-cl-20重结晶产物,保温10min后冷却到室温,抽滤,用40℃的蒸馏水洗涤3次,放入冷冻干燥机干燥。
[0062]
如图1所示,为本发明实施例1-9中go与tagn的化学键结合机理。
[0063]
实施例2
[0064]
一种石墨烯增韧二维高氮材料掺杂ε-cl-20晶体的制备方法,包括如下工艺步骤:
[0065]
氧化石墨烯悬浮液的配制:
[0066]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0067]
配制edc与nhs的去离子水溶液:
[0068]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0069]
go-tag的制备:
[0070]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0071]
以go-tag的掺杂量为1wt%,cl-20与go-tag的摩尔比为3:1,所加入tagn与乙二醛溶液摩尔比为1:1进行掺杂,先将23.3mg go-tag分散于10ml dmso中(在所需产量不同的情
况下,dmso的加入量为刚好使cl-20溶解为宜),体系温度保持在70~75℃,在磁力搅拌下得到go-tag的dmso分散液;
[0072]
再向上述混合液中加入250mg的tagn,磁力搅拌10min使混合更加均匀,再加入1972.5mgε-cl-20粉末,待加入的cl-20完全溶解后,缓慢滴加3滴40%乙二醛溶液,反应60min;
[0073]
将约10ml去离子水缓慢滴加到上述混合液中(蠕动泵的转速为150r/min),边滴加边搅拌,反应温度保持在70~75℃,得到以go-tagp掺杂ε-cl-20重结晶产物,保温10min后冷却到室温,抽滤,用40℃的蒸馏水洗涤3次,放入冷冻干燥机干燥。
[0074]
将实施例2制备的go-tagp掺杂ε-cl-20晶体进行扫描电子显微镜(sem)和透射电子显微镜(tem)分析,其晶体形貌如图2所示。由图2可知本实施例2制备的go-tagp掺杂ε-cl-20晶体中石墨烯片层分布均匀,go-tagp掺杂ε-cl-20晶体粒径约为8μm,且为立方体形状。
[0075]
将本实施例2制备的go-tagp掺杂ε-cl-20晶体进行热分析,分析结果如图9所示:该晶体热稳定性较好,转晶吸热峰温为202.4℃,相比于纯ε-cl-20提高了21.4℃,表明掺杂物的加入提高了ε-cl-20晶体的热稳定性。其热分解峰温为240.5℃,且热分解峰的总放热量为2433j/g,密度为1.965g/cm3,在提高热稳定性的同时保证了能量密度减小幅度较低。
[0076]
将本实施例2制备的go-tagp掺杂ε-cl-20晶体进行xrd晶型分析,分析结果如图10所示:实施例2和三种晶型(α-,β-,ε-)cl-20的xrd图在13.7时都存在较高的峰,它与ε和β晶型差距较大,但与α晶型差距小,仅在2θ=7.4和18.8时出现新的峰,因此可能出现新的晶型或晶相。
[0077]
实施例3
[0078]
一种石墨烯增韧二维高氮材料掺杂ε-cl-20晶体的制备方法,包括如下工艺步骤:
[0079]
氧化石墨烯悬浮液的配制:
[0080]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0081]
配制edc与nhs的去离子水溶液:
[0082]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0083]
go-tag的制备:
[0084]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0085]
以go-tag的掺杂量为1.5wt%,cl-20与go-tag的摩尔比为3:1,所加入tagn与乙二醛溶液摩尔比为1:1进行掺杂,先将35.2mg go-tag分散于10ml dmso中(在所需产量不同的情况下,dmso的加入量为刚好使cl-20溶解为宜),体系温度保持在70~75℃,在磁力搅拌下得到go-tag的dmso分散液;
[0086]
再向上述混合液中加入250mg的tagn,磁力搅拌10min使混合更加均匀,再加入1972.5mgε-cl-20粉末,待加入的cl-20完全溶解后,缓慢滴加3滴40%乙二醛溶液,反应60min;
[0087]
将约10ml去离子水缓慢滴加到上述混合液中(蠕动泵的转速为150r/min),边滴加
边搅拌,反应温度保持在70~75℃,得到以go-tagp掺杂ε-cl-20重结晶产物,保温10min后冷却到室温,抽滤,用40℃的蒸馏水洗涤3次,放入冷冻干燥机干燥。
[0088]
实施例4
[0089]
一种石墨烯增韧二维高氮材料掺杂ε-cl-20晶体的制备方法,包括如下工艺步骤:
[0090]
氧化石墨烯悬浮液的配制:
[0091]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0092]
配制edc与nhs的去离子水溶液:
[0093]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0094]
go-tag的制备:
[0095]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0096]
以go-tag的掺杂量为1wt%,cl-20与go-tag的摩尔比为4:1,所加入tagn与乙二醛溶液摩尔比为1:1进行掺杂,先将24mg go-tag分散于10ml dmso中(在所需产量不同的情况下,dmso的加入量为刚好使cl-20溶解为宜),体系温度保持在70~75℃,在磁力搅拌下得到go-tag的dmso分散液;
[0097]
再向上述混合液中加入200mg的tagn,磁力搅拌10min使混合更加均匀,再加入2106mgε-cl-20粉末,待加入的cl-20完全溶解后,缓慢滴加3滴40%乙二醛溶液,反应60min;
[0098]
将约10ml去离子水缓慢滴加到上述混合液中(蠕动泵的转速为150r/min),边滴加边搅拌,反应温度保持在70~75℃,得到以go-tagp掺杂ε-cl-20重结晶产物,保温10min后冷却到室温,抽滤,用40℃的蒸馏水洗涤3次,放入冷冻干燥机干燥。
[0099]
将实施例4制备的go-tagp掺杂ε-cl-20晶体进行扫描电子显微镜(sem)分析,其晶体形貌如图3所示。由图3可知本实施例4制备的go-tagp掺杂ε-cl-20晶体中颗粒表面光滑,与纯ε-cl-20相比形貌发生了较大变化,在结晶过程中晶面生长速度不一,导致晶体为扁平的立方体。
[0100]
实施例5
[0101]
一种石墨烯增韧二维高氮材料掺杂ε-cl-20晶体的制备方法,包括如下工艺步骤:
[0102]
氧化石墨烯悬浮液的配制:
[0103]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0104]
配制edc与nhs的去离子水溶液:
[0105]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0106]
go-tag的制备:
[0107]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0108]
制备步骤略。
[0109]
将实施例5制备的go-tagp掺杂ε-cl-20晶体进行扫描电子显微镜(sem)及透射电子显微镜(tem)分析,其晶体形貌如图4所示。由图4可知本实施例5制备的go-tagp掺杂ε-cl-20晶体使所得产品的表面光滑度下降,且晶体尺寸差异较大,表面形态多样,掺杂所使用的go为片层结构。
[0110]
将本实施例5制备的go-tagp掺杂ε-cl-20晶体进行xps化学键分析,分析结果为:以o1s吹扫时,o-n键含量为21.93%,o=n键含量为21.15%,c-o-c键含量为49.91%,o-h键含量为7.01%,以n1s吹扫时,n-n键含量为26.25%,n-h键含量为37.25%,n-o含量为17.61%,n-c含量为18.89%。由于xps进行表面10nm深度的元素检测,纯ε-cl-20的化学键峰位与go-tagp掺杂ε-cl-20晶体有部分差异,这可能是由于由纯cl-20表面包覆了go-tagp,xps测试结果检测到go-tagp中的化学键。
[0111]
将本实施例5制备的go-tagp掺杂ε-cl-20晶体进行热分析,分析结果如图9所示:该晶体热稳定性较好,且不存在转晶吸热峰,完全抑制了转晶的发生,表明以go-tagp掺杂晶体的热稳定性。其热分解峰温为244.0℃,热分解峰的总放热量为2115j/g,且其密度达到2.1244g/ml,在提高热稳定性的同时提高了密度。
[0112]
将本实施例5制备go-tagp掺杂ε-cl-20晶体进行xrd晶型分析,分析结果如图10所示:实施例5的xrd曲线与ε和β晶型差距较大,与α晶型在2θ=7.4和18.8时峰有微小区别,表明以溶剂-非溶剂法重结晶掺杂go-tagp的cl-20的晶体内原子排布发生了变化。
[0113]
实施例6
[0114]
一种石墨烯增韧二维高氮材料掺杂ε-cl-20晶体的制备方法,包括如下工艺步骤:
[0115]
氧化石墨烯悬浮液的配制:
[0116]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0117]
配制edc与nhs的去离子水溶液:
[0118]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0119]
go-tag的制备:
[0120]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0121]
以go-tag的掺杂量为1wt%,cl-20与go-tag的摩尔比为6:1,所加入tagn与乙二醛溶液摩尔比为1:1进行掺杂,先将17.3mg go-tag分散于10ml dmso中(在所需产量不同的情况下,dmso的加入量为刚好使cl-20溶解为宜),体系温度保持在70~75℃,在磁力搅拌下得到go-tag的dmso分散液;
[0122]
再向上述混合液中加入100mg的tagn,磁力搅拌10min使混合更加均匀,再加入1579mgε-cl-20粉末,待加入的cl-20完全溶解后,缓慢滴加2滴40%乙二醛溶液,反应60min;
[0123]
将约10ml去离子水缓慢滴加到上述混合液中(蠕动泵的转速为150r/min),边滴加边搅拌,反应温度保持在70~75℃,得到以go-tagp掺杂ε-cl-20重结晶产物,保温10min后冷却到室温,抽滤,用40℃的蒸馏水洗涤3次,放入冷冻干燥机干燥。
[0124]
将实施例6制备的go-tagp掺杂ε-cl-20晶体进行扫描电子显微镜(sem)分析,其晶
体形貌如图5所示。由图5可知本实施例6制备的以6:1掺杂比例的go-tagp掺杂ε-cl-20晶体使所得产品的表面有go-tagp包覆,晶体表面光滑。
[0125]
实施例7
[0126]
一种石墨烯增韧二维高氮材料掺杂ε-cl-20晶体的制备方法,包括如下工艺步骤:
[0127]
氧化石墨烯悬浮液的配制:
[0128]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0129]
配制edc与nhs的去离子水溶液:
[0130]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0131]
go-tag的制备:
[0132]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0133]
以go-tag的掺杂量为1wt%,cl-20与go-tag的摩尔比为7:1,所加入tagn与乙二醛溶液摩尔比为1:1进行掺杂,先将20mg go-tag分散于10ml dmso中(在所需产量不同的情况下,dmso的加入量为刚好使cl-20溶解为宜),体系温度保持在70~75℃,在磁力搅拌下得到go-tag的dmso分散液;
[0134]
再向上述混合液中加入100mg的tagn,磁力搅拌10min使混合更加均匀,再加入1842mgε-cl-20粉末,待加入的cl-20完全溶解后,缓慢滴加1滴40%乙二醛溶液,反应60min;
[0135]
将约10ml去离子水缓慢滴加到上述混合液中(蠕动泵的转速为150r/min),边滴加边搅拌,反应温度保持在70~75℃,得到以go-tagp掺杂ε-cl-20重结晶产物,保温10min后冷却到室温,抽滤,用40℃的蒸馏水洗涤3次,放入冷冻干燥机干燥。
[0136]
将实施例7制备的go-tagp掺杂ε-cl-20晶体进行扫描电子显微镜(sem)分析,其晶体形貌如图6所示。由图6可知本实施例7制备的go-tagp掺杂ε-cl-20晶体与纯ε-cl-20相似,为纺锤形,且表面光滑。
[0137]
实施例8
[0138]
一种石墨烯增韧二维高氮材料掺杂ε-cl-20晶体的制备方法,包括如下工艺步骤:
[0139]
氧化石墨烯悬浮液的配制:
[0140]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0141]
配制edc与nhs的去离子水溶液:
[0142]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0143]
go-tag的制备:
[0144]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0145]
以go-tag的掺杂量为1wt%,cl-20与go-tag的摩尔比为8:1,所加入tagn与乙二醛溶液摩尔比为1:1进行掺杂,先将22.6mg go-tag分散于10ml dmso中(在所需产量不同的情
况下,dmso的加入量为刚好使cl-20溶解为宜),体系温度保持在70~75℃,在磁力搅拌下得到go-tag的dmso分散液;
[0146]
再向上述混合液中加入100mg的tagn,磁力搅拌10min使混合更加均匀,再加入2105mgε-cl-20粉末,待加入的cl-20完全溶解后,缓慢滴加1滴40%乙二醛溶液,反应60min;
[0147]
将约10ml去离子水缓慢滴加到上述混合液中(蠕动泵的转速为150r/min),边滴加边搅拌,反应温度保持在70~75℃,得到以go-tagp掺杂ε-cl-20重结晶产物,保温10min后冷却到室温,抽滤,用40℃的蒸馏水洗涤3次,放入冷冻干燥机干燥。
[0148]
将实施例8制备的go-tagp掺杂ε-cl-20晶体进行扫描电子显微镜(sem)及透射电子显微镜(tem)分析,其晶体形貌如图7所示。由图7可知本实施例8制备的以8:1掺杂比例的go-tagp掺杂ε-cl-20晶体使所得产品的表面光滑度得到提高,但晶体尺寸差异较大,tem图显示掺杂所使用的go为片层结构。
[0149]
将本实施例8制备的go-tagp掺杂ε-cl-20晶体进行热分析,分析结果如图9所示:在热分析过程中该晶体未发生转晶,其热分解峰温为243.4℃(纯ε-cl-20为239.4℃),且热分解峰总放热量为2889j/g(纯ε-cl-20为2489j/g),且密度为2.0139g/ml,表明掺杂摩尔比例为8:1时可在保证能量密度的同时提高cl-20的热稳定性。
[0150]
将本实施例8制备的go-tagp掺杂ε-cl-20晶体进行xrd晶型分析,分析结果如图10所示:实施例8为cl-20与tagp摩尔比为8:1时所得产品,它的晶型与α-cl-20较为相似,但在2θ=7.4和18.8时,存在一些微小差异,与其dsc综合分析,推测可能出现了新的晶型。
[0151]
实施例9
[0152]
一种石墨烯增韧二维高氮材料掺杂β-hmx晶体的制备方法,包括如下工艺步骤:
[0153]
氧化石墨烯悬浮液的配制:
[0154]
将go称量20mg加入10ml水(也可以是其它有机溶剂,如乙醇、丙醇、二甲基甲酰胺、二甲基亚砜等)中,超声分散60min可得到氧化石墨烯悬浮液(配置浓度为2mg/ml);
[0155]
配制edc与nhs的去离子水溶液:
[0156]
将4mg edc和3mg nhs加入到3ml的去离子水中,摇匀溶解;
[0157]
go-tag的制备:
[0158]
在70~75℃温度下,将所得edc与nhs去离子水溶液加入到go悬浮液中,在此温度下反应30min,得到活化羧基的go分散液;再将200mg tagn加入到上述混合液中,反应4h,得到go-tag的黑色絮状沉淀,自然冷却到室温后抽滤干燥。
[0159]
以go-tag的掺杂量为1wt%,hmx与go-tag的摩尔比与最优的cl-20掺杂物相同,所加入tagn与乙二醛溶液摩尔比为1:1进行掺杂,先将20.5mg go-tag分散于10ml dmso中(在所需产量不同的情况下,dmso的加入量为刚好使hmx溶解为宜),体系温度保持在70~75℃,在磁力搅拌下得到go-tag的dmso分散液;
[0160]
再向上述混合液中加入200mg的tagn,磁力搅拌10min使混合更加均匀,再加入1768mgβ-hmx粉末,待加入的hmx完全溶解后,缓慢滴加2滴40%乙二醛溶液,反应60min;
[0161]
将约10ml去离子水缓慢滴加到上述混合液中(蠕动泵的转速为150r/min),边滴加边搅拌,反应温度保持在70~75℃,得到以go-tagp掺杂β-hmx重结晶产物,保温10min后冷却到室温,抽滤,用40℃的蒸馏水洗涤3次,放入冷冻干燥机干燥。
[0162]
将实施例9制备的go-tagp掺杂β-hmx晶体进行扫描电子显微镜(sem)分析,其晶体形貌如图8所示。由图8可知,本实施例9制备的go-tagp掺杂β-hmx晶体使所得产品的表面光滑度得到提高,晶体尺寸分布均匀,与纯hmx的外观形貌较为相似。
[0163]
将本实施例9制备的go-tagp掺杂β-hmx晶体进行热分析,分析结果如图9所示:该晶体转晶峰温为197.9℃(纯β-hmx为199.4℃),热分解峰温为282.3℃(纯β-hmx为279.8℃),且热分解峰总放热量为1635j/g(纯β-hmx为1823j/g),且密度为1.8609g/ml。
[0164]
将本实施例9制备的go-tagp掺杂β-hmx晶体进行xrd晶型分析,分析结果如图10所示:实施例9为hmx与tagp摩尔比最优的cl-20掺杂物相同的产品,它的晶型与β-hmx相同,各个峰强度有所改变,但其晶型与β-hmx保持一致。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips