
 商标分类
商标分类  商标转让
商标转让 
新型金属层状氢氧化物复合物及其制备方法与流程
 2021-01-08 11:01:01|
2021-01-08 11:01:01| 261|
261| 起点商标网
起点商标网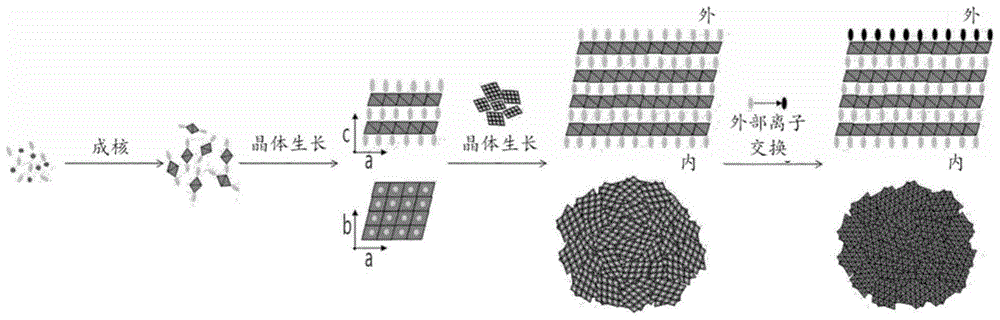
本发明涉及一种包括活性成分和肽类表面改性剂的新型金属层状氢氧化物复合物及其制备方法。
背景技术:
:为了改善常规金属层状氢氧化物复合物等的受控释放性(controlledreleasability)并调节其分子尺寸,已经使用热处理、超声处理或重建对其执行了表面处理。然而,这些表面处理方法是有问题的,因为可能发生活性成分的分解和活性成分的意外释放,并且还因为需要长的处理时间及其制备设施。此外,以减轻上述方法中需要制备设施的问题为目的,聚合物吸附法或分散剂处理法可以被应用,但是具有长的处理时间,并且根据活性成分、聚合物或分散剂的类型可能会使用有机溶剂,因此使用受到限制。此外,主要合成后执行后处理工艺,其效率低下,特别地,缺点是洗涤期间产率可能降低。因此,金属层状氢氧化物的表面改性通过分层或剥离工艺进行,该分层或剥离工艺通过溶胀来分离纳米片。然而,为此目的,必须使用有机溶剂,诸如甲苯、甲醛、二氯甲烷等,这需要诸如洗涤等的后处理工艺,并且引起生物稳定性问题。此外,纳米片的完全分离使得不可能通过将不稳定的活性成分引入层状结构中来稳定该不稳定的活性成分。类似地,尽管在金属层的表面上施用了使用诸如氨基硅烷、环氧硅烷或乙烯基硅烷的配体结合官能团的方法,但是由于有机溶剂的使用,该方法也引起了上述问题。具体地,在复合物的洗涤和干燥之后,由于氢键合和干燥,颗粒的表面经受了在纳米片之间共享水分子的物理现象,因此颗粒可能会聚集,因此颗粒尺寸最终会更大。此外,在洗涤和干燥后对复合材料进行表面处理时,由于表面边缘效应(surfaceedgingeffect),形成未活化的表面层,因此颗粒之间的相互作用稳定,且阴离子键合或分散非常困难,因此整体表面改性效率可能会恶化,这是不期望的。通常,如韩国专利第10-0439299号和第10-0841700号中公开的用作表面改性剂的teos或tmos主要由二氧化硅组成,因此表现出受控的释放性,但是由于二氧化硅的性质,teos或tmos具有高的水含量,这会不期望地引起与活性成分的稳定性有关的问题。另外,当使用韩国专利申请公开第10-2006-0132409号中公开的卵磷脂、纤维素、黄原胶、瓜尔胶、甲基纤维素和羟丙基甲基纤维素(hydroxypropylmethylcellulose)时,金属层状氢氧化物和改性剂仅以简单混合的形式提供,而不是物理和化学结合,因此不会出现颗粒尺寸控制和分散性能,这是不希望的。作为上述常规表面处理方法,通过约100℃下的处理和超声处理来蚀刻颗粒表面以改变颗粒表面的物理性能的高温热处理或超声处理方法、通过200℃以上的高温下的处理诱导颗粒结构变化的重建方法、将聚合物吸附至颗粒表面的聚合物或分散剂吸附方法、使用有机溶剂剥离金属层状氢氧化物的方法、以及使用官能团表面改性的方法全部都是有问题的,因为需要制备设施或制备工艺复杂,并由于有害溶剂的使用而引起安全问题,因此,需要替代的改性方法。因此,本发明旨在提供一种新型的金属层状氢氧化物复合物,其不具有前述问题并且是表面改性的。(专利文献1)韩国专利第10-0439299号(专利文献2)韩国专利第10-0841700号(专利文献3)韩国专利申请公开第10-2006-0132409号技术实现要素:技术问题做出本发明以针对相关技术中遇到的问题而做出的,并且本发明的目的是提供一种金属层状氢氧化物复合物,其包括活性成分和表面改性剂、且具有减小的颗粒尺寸,其中稳定地含有活性成分,并且其受控的释放性显著改善。本发明的另一个目的是提供一种制备金属层状氢氧化物复合物的方法,其中金属层状氢氧化物复合物可以以如下方式简单地合成:使金属层状氢氧化物通过静电吸引结合至具有相反电荷的活性成分,或者调节金属层状氢氧化物的内部结构,即其结晶度,从而使得可以在不使用有害材料的情况下安全地制备金属层状氢氧化物复合物,实现了最大的合成效率和短的处理时间,并产生了环境友好和经济的效果。技术方案本发明提供了一种包括活性成分和表面改性剂的金属层状氢氧化物复合物,该活性成分为选自由以下组成的组的至少之一:抗坏血酸、生物素、泛酸、半胱氨酸、阿魏酸、谷氨酸、吲哚乙酸、肉桂酸(cinnamicacid)、咖啡酸、硫胺素(thiamine)、核黄素、烟酸、泛酸、吡哆素(pyridoxine)、叶酸、钙化醇、生育酚、氨甲环酸(tranexamicacid)、水杨酸、视黄酸、和熊果苷,并且该表面改性剂包括选自由以下组成的组的至少之一:含有2至8个氨基酸的肽、以及包括含有2至8个氨基酸的肽和含有10至16个碳原子的脂肪酸的肽/脂肪酸偶联物(peptide/fatty-acidconjugate)。另外,本发明提供了一种制备上述金属层状氢氧化物复合物的方法,该方法包括:通过将酸性溶液逐滴地添加到金属层状氢氧化物复合物的前体并溶解该前体来制备前体溶液;以及通过将前体溶液与醇、去离子水、活性成分和表面改性剂混合来制备金属层状氢氧化物复合物。另外,本发明提供了一种制备上述金属层状氢氧化物复合物的方法,该方法包括:通过将酸性溶液逐滴地添加到金属层状氢氧化物复合物的前体并溶解该前体来制备前体溶液;通过将前体溶液与醇、去离子水和活性成分混合来制备含有活性成分的晶种(crystalseed);以及通过将所制备的晶种与表面改性剂混合并执行离子交换反应来制备金属层状氢氧化物复合物,其中离子交换反应中的ph为5至10。有利效果根据本发明,提供了一种包括活性成分和表面改性剂的金属层状氢氧化物复合物,由于减小的颗粒尺寸、活性成分的稳定内含物(inclusion)以及极好的活性成分的受控释放性,该金属层状氢氧化物复合物是有利的。此外,根据本发明,金属层状氢氧化物复合物可以以如下方式简单地合成:使金属层状氢氧化物通过静电吸引结合至具有相反电荷的活性成分,或者调节金属层状氢氧化物的内部结构、即其结晶度,从而实现短的处理时间,并省去了专用的制备设施,因此产生了环境友好和经济的效果。另外,根据本发明,制备金属层状氢氧化物复合物的方法省略了由于使用诸如甲苯、甲酰胺或二氯甲烷的有机溶剂引起的诸如洗涤等的后处理,不使用有害材料,并因此是安全的。附图说明图1示出了使用外部离子交换工艺(outerino-exchangeprocess)形成金属层状氢氧化物复合物的工艺;图2示出了使用外部离子交换工艺形成的金属层状氢氧化物复合物。图3示出了使用交叉插入工艺(crossoverinterpositionprocess)形成金属层状氢氧化物复合物的工艺;图4示出了使用交叉插入工艺形成的金属层状氢氧化物复合物;图5-图10是金属层状氢氧化物复合物的xrd图谱;图11-图20是示出了金属层状氢氧化物复合物的丁达尔效应的照片;图21示出了使用外部离子交换工艺制备的金属层状氢氧化物复合物(其中,其结晶度在sma0.005∶vitc1的比例下来调节)的内-外结构;图22示出了使用交叉插入工艺制备的金属层状氢氧化物复合物(其中,其结晶度在sma0.5:vitc0.5的比例下来调节)的内-外结构;图23和图24是示出了金属层状氢氧化物复合物的颗粒尺寸的评估结果的图;图25和图26是示出了金属层状氢氧化物复合物的zeta电位的评估结果的图;图27和图28是示出了金属层状氢氧化物复合物的颗粒尺寸的评估结果的图;图29和图30是示出了金属层状氢氧化物复合物的zeta电位的评估结果的图;以及图31-图35是示出了金属层状氢氧化物复合物的受控释放性的评估结果的图。最佳方式本发明涉及一种包括活性成分和表面改性剂的新型金属层状氢氧化物复合物、及其制备方法。根据插入其中的材料的类型(例如表面活性剂、氨基酸等),金属层状氢氧化物复合物可用作功能材料或药物材料,诸如口服剂、清洁剂或洗发剂,并且在保持活性成分的稳定性和使胶体形式的颗粒尺寸变小方面有效,从而使得其在用作化妆品时可以增加其吸收能力、并表现出优异的活性成分的受控释放性。具体地,根据本发明的金属层状氢氧化物复合物的受控释放性使得活性成分在72小时内以65%以下的释放率释放,并且优选在72小时内以30-60%的释放率释放。此外,本发明的金属层状氢氧化物复合物的颗粒尺寸(z平均(d.nm))小于1000d.nm,且优选为200-900d.nm。在下文中,将给出本发明的构造的详细描述。<新型的金属层状氢氧化物复合物>在本发明中,金属层状氢氧化物复合物可以是金属和氢氧化物彼此结合的复合物。优选地,其由以下化学式1表示,但不限于此。[化学式1](在化学式1中,m2+为mg2+、ca2+、co2+、cu2+、ni2+或zn2+,a为带负电的生物活性材料,x′为小于2但超过1的数字;y为100以下但超过0的数字;并且w为0或1)。在本发明中,仅以区分活性成分和表面改性剂的方式命名活性成分和表面改性剂,表面改性剂可以用作活性成分,并且活性成分也可以用作表面改性剂。在本发明中,活性成分可以是选自由以下组成的组的至少之一:抗坏血酸、生物素、泛酸、半胱氨酸、阿魏酸、谷氨酸、吲哚乙酸、肉桂酸、咖啡酸、硫胺素、核黄素、烟酸、泛酸、吡哆素、叶酸、钙化醇、生育酚、氨甲环酸、水杨酸、视黄酸和熊果苷。在本发明中,表面改性剂可以包括选自由以下组成的组的至少之一:含有2至8个氨基酸的肽、以及包括含有2至8个氨基酸的肽和含有10至16个碳原子的脂肪酸的肽/脂肪酸偶联物。在本发明中,构成各肽的缩写一般对应于通常用于表达氨基酸的缩写。具体地,在本发明中,含有2至8个氨基酸的肽可以是选自由以下组成的组的至少之一:二肽-1(yr)、二肽-2(vw)、二肽-4(fw)、二肽-6(kv)、二肽-7(kt)、二肽-14(at)、二肽(gh)、乙酰基二肽-1(yr)、乙酰基二肽-1鲸蜡酯(yr)、烟酰基二肽-2(vw)、cp二肽(cp)、vge二肽(ve)、cge二肽(ce)、ege二肽(ee)、tge二肽(te)、lge二肽(le)、eq二肽(eq)、gr二肽(gr)、hg二肽(hg)、pe二肽(pe)、de二肽(de)、hq二肽(hq)、rs二肽(rs)、hp二肽(hp)、肌肽(ah)、三肽-1(ghk)、三肽-3(ghr)、三肽-4(lgd)、三肽-5(kvk)、三肽-6(gxp)、三肽-8(hfr)、三肽-10(kdi)、rgd肽(rgd)、ahk肽(ahk)、三肽-29(gpx)、三肽-54(fty)、生物素三肽-1(biotinoyltripeptide-1)(ghk)、硫代辛酰基基三肽-1(thioctoyltripeptide-1,ghk)、三肽(rfk)、hgg肽(hgg)、肽ck(rkr)、四肽-1(lptv)、四肽-2(kdvy)、四肽-3(kghk)、四肽-5(ahsh)、四肽-7(gqpr)、四肽-9(qdvh)、四肽-11(ppyl)、四肽-15(ypff)、四肽-21(gekg)、四肽-26(elps)、乙酰基四肽-2(kdvy)、乙酰基四肽-3(kghk)、乙酰基四肽-5(ahsh)、乙酰基四肽-9(qdvh)、乙酰基四肽-11(ppyl)、乙酰基四肽-15(ypff)、五肽-3(gprpa)、五肽-4(kttks)、五肽-17(klakk)、五肽-18(yagfl)、硫代辛酰基基五肽-4(thioctoylpentapeptide-4,kttks)、六肽-1(arhlfw)、六肽-2(fwfkpv)、六肽-3(eemqrr)、六肽-4(fghxaf)、六肽-5(fgvxaf)、六肽-6(vepipy)、六肽-9(gpqgpq)、六肽-11(fvapfp)、六肽-12(vgvapg)、乙酰基六肽-1(arhlfw)、乙酰基六肽-3(eemqrr)、乙酰基六肽-6(vepipy)、七肽-1(edddwdf)、七肽-7(mgrnirn)、半胱氨酸肽(rfaacaa)、赖氨酸肽(rfaakaa)、西蓝科(selank)(tkprpgp)、八肽-2(taeehevm)、八肽-3(eemqrrad)、八肽-4(yggflghk)和乙酰基八肽-3(eemqrrad)。在本发明中,肽/脂肪酸偶联物可以是选自由以下组成的组的至少之一:棕榈酰二肽-6(kv)、棕榈酰二肽-7(kt)、棕榈酰肌肽(ah)、壬二酰三肽-1(ghk)、棕榈酰三肽-3(ghr)、肉豆蔻酰三肽-5(kvk)、棕榈酰三肽-1(ghk)、棕榈酰三肽-5(kvk)、棕榈酰三肽(rfk)、肉豆蔻酰三肽-1(ghk)、棕榈酰三肽-4(lgd)、棕榈酰三肽-8(hfr)、棕榈酰四肽-7(gqpr)、肉豆蔻酰五肽-17(klakk)、棕榈酰五肽-4(kttks)、棕榈酰五肽-17(klakk)、肉豆蔻酰六肽-12(vgvapg)和棕榈酰六肽-12(vgvapg)。在本发明中,根据活性成分和表面改性剂的类型,可以将新型的金属层状氢氧化物复合物应用于化妆品、清洁剂、药物等。特别地,所制备的材料的功能性可以根据用作活性成分和/或表面改性剂的阴离子的性能而变化。具体地,使用抗坏血酸作为活性成分和表面改性剂制备的产品可以用作化妆品,其在应用于皮肤时具有改善的吸收能力,或者使用抗坏血酸作为活性成分和表面活性剂制备的产品可以作为具有抗氧化功效的清洁剂而起作用。另外,本发明的金属层状氢氧化物复合物可以用作化妆品或药物组合物。按照传统工艺,包括本发明的金属层状氢氧化物复合物的药物组合物可以进一步包括适当的载体、赋形剂或稀释剂。可以包括在含有化合物的药物组合物中的载体、赋形剂和稀释剂的实施例可以包括但不限于:乳糖、右旋糖、蔗糖、山梨糖醇、甘露醇、木糖醇、赤藓糖醇、麦芽糖醇、淀粉、阿拉伯橡胶(acaciarubber)、藻酸盐、明胶、磷酸钙、硅酸钙、纤维素、甲基纤维素、微晶纤维素、聚乙烯吡咯烷酮、水、对羟基苯甲酸甲酯(methylhydroxybenzoate)、对羟基苯甲酸丙酯(propylhydroxybenzoate)、滑石粉、硬脂酸镁和矿物油。另外,按照传统工艺,本发明的药物组合物可以配制成选自由以下组成的组的任何一种剂型:散剂、丸剂、颗粒剂、胶囊、悬浮剂、内用溶液、乳剂、糖浆、无菌水溶液、非水溶液、冻干剂和栓剂。当配制本发明的药物组合物时,可以使用传统的稀释剂或赋形剂,诸如填充剂、增量剂、粘合剂、润湿剂、崩解剂和表面活性剂等。用于口服给药的固体制剂可以包括片剂、丸剂、散剂、颗粒剂和胶囊等,并且这种固体制剂可以通过将上述化合物与至少一种赋形剂混合来制备,该赋形剂例如淀粉、碳酸钙、蔗糖、乳糖或明胶等。除了简单的赋形剂外,可以使用润滑剂,诸如硬脂酸镁和滑石粉等。口服液体制剂可以包括悬浮剂、内用溶液、乳剂和糖浆等,并且不仅可以包括简单的稀释剂,诸如水或液体石蜡,还可以包括各种赋形剂,例如润湿剂、甜味剂、香料和防腐剂等。用于肠胃外给药的制剂可以包括无菌水溶液、非水溶剂、悬浮剂、乳剂、冻干制剂和栓剂。作为非水溶剂或悬浮剂,可以使用丙二醇、聚乙二醇、诸如橄榄油的植物油、以及诸如油酸乙酯的可注射酯等。作为栓剂的基质,可以使用合成脂肪酸酯(witepsol)、聚乙二醇(macrogol)、吐温60(tween60)、可可脂、月桂脂和甘油明胶(glycerogelatin)等。包括本发明的金属层状氢氧化物复合物的药物组合物可以根据使用目的而口服给药或胃肠外(例如,静脉内、皮下、腹膜内或局部)给药;并且在给药时,根据本发明的药物组合物的量可以根据患者的健康状况、体重、年龄、性别、饮食、排泄率、疾病严重性、药物形式、给药时间、给药途径和给药周期而变化,并且可以由本领域技术人员适当地确定。因此,本发明的范围不以任何方式受到限制。本发明的化妆品或药物组合物可以具体地是用于外用至皮肤的制剂,并且更具体地,用于外用至皮肤的制剂可以包括选自由以下组成的组的任一种:溶液、悬浮剂、乳剂、糊剂、凝胶剂、霜剂、洗剂、散剂、肥皂、含表面活性剂的清洁剂、油、粉底、乳液粉底、蜡粉底和喷雾剂,但不限于此,只要该制剂是用于应用至皮肤的制剂即可。<制备金属层状氢氧化物复合物的方法>本发明的金属层状氢氧化物复合物可以使用外部离子交换工艺或交叉插入工艺来制备,但本发明不限于此。根据本发明的制备金属层状氢氧化物的方法使得可以控制颗粒尺寸并表现出优异的分散性。上述方法是通过在合成开始后允许由成核而产生的晶种的最大聚集生长时间来将颗粒生长至最大尺寸来的表面改性的方法,并且从以下观点出发上述方法是优选的:由于洗涤之前水溶液中颗粒之间的结合力弱于洗涤和干燥后(因为水溶液中存在不同的抗衡阴离子或金属盐),所以颗粒尺寸较小且颗粒之间的表面积较大,因此表面处理是非常有用的。使用阴离子材料之间的性能来执行颗粒的表面改性,该阴离子材料含有具有取决于pka的静电吸引力的官能团(官能团:羟基(-oh)、酮的羰基(-co-)、醛的甲酰基(-cho)、有机酸的羧基(-cooh)、胺的氨基(-nh2)、硫酸盐、磷酸盐等),该官能团的活性在水溶液中非常高。在本发明中,金属层状氢氧化物复合物的前体的具体实施例可以包括zno、znso4、zncl2、znco3、zn(no3)-6h2o、zn(ch3coo)2、cao、cacl2、ca(no3)26h2o、caso4、caco3、ca(oh)2、mgo、mgcl2、mgso4、mg(no3)26h2o、cuo、cu(no3)26h2o、cucl2、cuso4、co(no3)2、coco3、cocl2、co(oh)2、coso4、co(ch3coo)2、nio、nicl2、ni(no3)2、niso4、nico3、ni(oh)2、feo、fecl3、fe(no3)3、feso4、feco3和fe(oh)2。优选地,使用zno、znso4、zncl2、znco3、zn(no3)26h2o、或zn(ch3coo)2。具体地,外部离子交换工艺可以包括:通过将酸性溶液逐滴地添加到金属层状氢氧化物复合物的前体并使该前体溶解于其中来制备前体溶液;通过将前体溶液与醇、去离子水、活性成分等混合来制备含有活性成分的晶种;以及通过将所制备的晶种与表面改性剂混合并执行离子交换反应来制备金属层状氢氧化物复合物。更具体地,外部离子交换工艺是一种合成方法,其中使结合到活性成分的种颗粒(seedparticle)的聚集生长时间最大化,从而增加预定时间段内的晶体的尺寸,然后在洗涤前使用表面改性剂在颗粒表面上进行离子交换。当离子交换反应中的ph的范围从5至10时,通过保持现有的、用于合成金属层状氢氧化物的ph条件,可以最小化由于ph变化而引起的活性成分的释放,此外,可以防止金属层状氢氧化物结构的坍塌,从而增加活性成分的受控释放性。至于在离子交换反应中使用的肽,优选具有5至10的pi的肽,其满足ph条件在5至10的范围内。在本发明中,pi(等电点)是指肽的静电性能,并且意味着由肽的pka的偶极结构的静电性能表示的平均值。pi与肽的pka密切相关;例如,含有一个氨基和一个羧基的氨基酸的pi可以由方程式(pka1+pka2)/2=pi表示,并且肽的pi值可以通过常用的pi计算方法而容易地得出。在5至10的ph处,具有pi值的肽具有适合于氢键合或羧酸物键合等的键合活性基团,因此可以在离子交换期间表现出优异的静电吸引。当制备本发明的金属层状氢氧化物复合物时,活性成分和表面改性剂可以以1:0.0005至0.0005:1的摩尔比混合。考虑到上述范围,可以有效地调节活性成分的受控释放性,此外,可以减小金属层状氢氧化物的颗粒尺寸,并且可以减少不必要的涂覆剂(coatingagent)的使用。更具体地,在离子交换反应中,使用阴离子交换量来进行表面改性。外部离子交换工艺能够形成一种配置为使得金属层状氢氧化物复合物的最外表面被表面改性剂层包围的结构(图1和图2)。另外,交叉插入工艺可以包括:通过通过将酸性溶液逐滴地添加到金属层状氢氧化物复合物的前体并将该前体溶解于其中来制备前体溶液;以及通过将前体溶液与醇、去离子水、活性成分和表面改性剂混合来制备金属层状氢氧化物复合物。当制备本发明的金属层状氢氧化物复合物时,活性成分和表面改性剂可以以0.9995:0.0005至0.0005:0.9995的摩尔比混合。交叉插入工艺能够形成一种配置为使得活性成分和表面改性剂以混合物的形式提供在金属层状氢氧化物复合物的内部和最外表面上的结构(图2)。发明方式下面描述根据本发明的具体制备方法。然而,本发明不限于以下实施例。制备实施例制备实施例1:使用外部离子交换工艺制备金属层状氢氧化物复合物作为实验的基本条件,所有工艺均在室温(20℃)和氮气气氛下执行。此外,使用95%的乙醇和去离子水,并使用纯度为95%以上(水含量为5%以下)的抗坏血酸(aa)和sma(表面改性剂)。使用去离子水制备3.2mnaoh水溶液。本发明中使用的表面改性剂sma1和sma2分别为五肽-4和棕榈酰二肽-7。实验按照合成、洗涤和干燥的顺序如下进行。将2.5当量的zno放在主槽(maintank)中,然后将辅助槽1中的浓hcl溶液逐滴地缓慢添加到主槽,从而将ph滴定至0.5-1,然后在氮气气氛下、在700rpm的搅拌下溶解约30分钟。此后,将辅助槽2中的1当量的aa、去离子水和乙醇放在主槽中(添加的溶剂相对于zn的总体积比为20),然后在300rpm的搅拌下溶解约5分钟。在保持主槽的搅拌速度的情况下,将辅助槽3中的3.2mnaoh溶液添加到主槽,从而将主槽中的ph滴定到6.5至7.5的范围内,然后搅拌3小时,在主槽中引起沉淀反应。这里,ph保持在6.5-7.5。接下来,洗涤沉淀物。具体地,为了去除上述溶液和离子材料中的未反应的盐,使用离心机去除杂质。离心工艺以8000rpm执行3次,每次5分钟,并且如下所述将沉淀物和溶液彼此分离。将合成的沉淀物溶液以8000rpm离心5分钟。分离溶液和沉淀物之后,将沉淀物用乙醇和去离子水以1:1的体积比均匀稀释,然后以8000rpm离心5分钟。该程序重复三次。在最后的离心洗涤工艺中,仅用乙醇来稀释沉淀物,然后以9000rpm离心10分钟,从而产生沉淀物。接下来,执行表面改性。具体地,将洗涤后分离出的沉淀物在氮气气氛中使用乙醇和去离子水以1:9至9:1的体积比稀释,然后以700rpm搅拌30分钟。向分散的沉淀物溶液缓慢地添加sma溶液,sma相对于aa以1:0.05、1:0.025、1:0.005、1:0.0025和1:0.0005的摩尔比溶解在乙醇中(50%),然后在使用弱碱性溶液将其ph保持在ph6-8的情况下搅拌约1至3小时。反应完成后,将沉淀物以8000rpm离心。在溶液与沉淀物彼此分离之后,将沉淀物用乙醇和去离子水以1:1的比例均匀稀释,然后以8000rpm离心5分钟。该程序重复三次。在最后的离心洗涤工艺中,仅用乙醇来稀释沉淀物,然后以9000rpm将沉淀物离心10分钟,从而产生沉淀物。在600rpm的搅拌下将洗涤的沉淀物溶液喷雾干燥,从而获得粉末。制备实施例2:使用交叉插入工艺制备金属层状氢氧化物复合物作为实验的基本条件,所有工艺均在室温(20℃)和氮气气氛下执行。此外,使用95%的乙醇和去离子水,并使用纯度为95%以上(水含量为5%以下)的抗坏血酸(aa)和sma(表面改性剂)。使用去离子水制备3.2mnaoh水溶液。本发明中使用的表面改性剂sma1和sma2分别为五肽-4和棕榈酰二肽-7。实验按照合成、洗涤和干燥的顺序如下进行。具体地,将2.5当量的zno放在主槽中,然后将辅助槽1中的浓hcl溶液逐滴地缓慢添加到主槽,从而将ph滴定至0.5-1,然后在氮气气氛下、在700rpm的搅拌下溶解约30分钟。此后,将辅助槽2中的1当量的aa和1当量的sma、去离子水和乙醇放在主槽中(添加的溶剂相对于zn的总体积比为20),然后在300rpm的搅拌下溶解约10分钟。此后,搅拌主槽,然后将辅助槽3中的3.2mnaoh溶液添加到主槽,从而将主槽中的ph滴定到6.5至7.5的范围内,然后搅拌3小时,在主槽中引起沉淀反应。这里,其ph保持在6.5-7.5。接下来,洗涤沉淀物。具体地,为了去除上述溶液和离子材料中的未反应的盐,使用离心机去除杂质。离心工艺以8000rpm执行3次,每次5分钟,并且如下所述将沉淀物和溶液彼此分离。将合成的沉淀物溶液以8000rpm离心5分钟。分离溶液和沉淀物之后,将沉淀物用乙醇和去离子水以1:1的体积比均匀稀释,然后以8000rpm离心5分钟。该程序重复三次。在最后的离心洗涤工艺中,仅用乙醇来稀释沉淀物,然后以9000rpm将沉淀物离心10分钟,从而产生沉淀物。在600rpm的搅拌下将洗涤的沉淀物溶液喷雾干燥,从而获得粉末。实施例和比较实施例实施例1-20和比较实施例1-4除组分的量如下表1和表2所示的改变之外,以与制备实施例1相同的方式制备金属层状氢氧化物复合物。[表1][表2]实施例21-34和比较实施例1-3除组分的量如下表3和表4所示的改变之外,以与制备实施例2相同的方式制备金属层状氢氧化物复合物。[表3][表4]试验性实施例1.xrd图谱的测量1-1.制备实施例1中制备的金属层状氢氧化物的xrd图谱和d值的测量分析方法1:粉末x射线衍射图-仪器:粉末x射线衍射(pxrd)x射线衍射仪(d/maxprint2200-ultima,日本,日本理学(rigaku))cu-kα辐射管电压:40kv,电流:30ma作为x射线衍射仪,使用可从日本理学(日本)获得的d/maxprint2200-ultima。产生x射线的阳极由cu金属形成,并且使用kα射线在3-70°的2θ处执行测量,扫描速度为0.02°/0.2秒,并且发散狭缝、散射狭缝和接收狭缝分别为0.1、1和1mm。管电压为40kv,电流为30ma。–评估标准使用以下方程式1计算z轴的一维(1d)电子密度。[方程式1]使用xrd图谱对通过合成获得的粉末进行比较分析,并且使用布拉格方程式(下面的方程式2)计算层间距离。对于最前面的峰,其表示包括合成的金属氢氧化物层和阴离子层之间的距离的层间距离,合成的金属氢氧化物层和阴离子层之间的距离被视为主要的层间距离。[方程式2]nλ=2dsinθ(λ=x射线波长,d=晶格间距,θ=入射角)使用与上述1-1相同的分析方法进行实验和评估。在表5和图5中,实施例1以样品①表示,实施例3以样品②表示,实施例5以样品③表示,并且在表6和图6中,实施例11以样品①表示,实施例13以样品②表示,实施例15以样品③表示。其具体值在表5和表6(图5和图6)中示出。另外,在表7和图7中,实施例6以样品(1)表示,实施例8以样品(2)表示,实施例10以样品(3)表示,并且在表8和图8中,实施例16以样品(1)表示,实施例18以样品(2)表示,实施例20以样品(3)表示。其具体值在表7和表8(图7和图8)中示出。参考实施例:锌层状氢氧化物的制备5克zn(no3)2·6h2o溶解在不含碳酸根离子(co32-)的去离子水中,然后使用0.2mnaoh滴定至ph约为6至7,从而获得锌碱性盐沉淀物。将由此滴定的溶液经受离心和洗涤,以去除未反应的盐,从而获得2.6g的白色粉末(产率:70%)。[表5][表6]从上表中可以明显看出,基于通过添加基于活性成分(抗坏血酸)比的sma1或sma2表面改性的结果,各样品的主峰的θ(θ)值在使用sma1时为5.9、5.92和5.94,在使用sma2时分别为6.04、6.06和5.96;并且d值(层间距离值)在使用sma1时为14.97、14.92和在使用sma2时为14.62、14.57和这些被确认为几乎一样。当使用外部离子交换工艺时,可以确认sma没有插入到层间间隔中,而是存在于表面上。[表7][表8]从上表中可以明显看出,基于在内组分为sma时、内组分的摩尔比为1的条件下通过添加抗坏血酸表面改性的结果,各样品的主峰的θ(θ)值在使用sma1时为2.25、2.25和2.37,在使用sma2时分别为2.54、2.59和2.53;并且d值(层间距离值)在使用sma1时为39.8、39.8和在使用sma2时为34.7、34.08和这些被确认为几乎一样。这表明aa没有插入到层间间隔中,而是存在于表面上。1-2.制备实施例2中制备的金属层状氢氧化物的xrd图谱和d值的测量在表9和图9中,比较实施例5以样品①表示,实施例21以样品②表示,实施例22以样品③表示,实施例23以样品④表示,并且在表10和图10中,比较实施例7以样品①表示,实施例28以样品②表示,实施例29以样品③表示,实施例30以样品④表示。其具体值在表9和表10(图9和图10)中示出。[表9]描述:在通过交叉插入工艺以不同比例使用sma1和vitc(抗坏血酸)合成后、通过控制结晶度的内-外表面处理后的xrd图谱。①sma11:vitc0②sma10.5:vitc0.5③sma10.25:vitc0.75④sma10:vitc1评估了用sma1的zlh-aa的内外表面处理后的xrd图谱变化。确认了插入到zlh中的阴离子的xrd峰的强度根据基于活性成分的sma1和vitc的比例变化而变化。当单独使用sma1(1)时,观察到值为2.25、4.43、6.61和8.83,这表明结晶度高且峰位对齐良好。这里,其层间间隔为当sma1和vitc的比例从样品1变到样品4时,层间距离发生变化,这表明代表位于层中的sma1的主峰的位置发生了变化,并且出现了逐渐减小层间距离的效果,同时产生了新的、大的、宽的vitc主峰,最终sma1峰消失。[表10]描述:在通过交叉插入工艺以不同比例使用sma2和vitc(抗坏血酸)合成后、通过控制结晶度的内-外表面处理后的xrd图谱。①sma21:vitc0②sma20.5:vitc0.5③sma20.25:vitc0.75④sma20:vitc1评估了用sma2的zlh-aa的内外表面处理后的xrd图谱变化。确认了插入到zlh中的阴离子的xrd峰的强度根据基于活性成分的sma2和vitc的比例变化而变化。当单独使用sma2(1)时,观察到值为2.59、5.21、7.73和10.42,这表明结晶度高且峰位对齐良好。这里,其层间间隔为当sma2和vitc的比例从样品1变到样品4时,层间距离发生变化,这表明代表位于层中的sma2的主峰的位置发生了变化,并且出现了逐渐减小层间距离的效果,同时产生了新的、大的、宽的vitc主峰,最终sma2峰消失。试验性实施例2.zn、sma1、sma2和aa的量的测量分析方法2:hplc分析-抗坏血酸仪器:高效液相色谱(hplc)分析agilent1100系列(美国安捷伦科技公司)紫外线检测器(λmax=240)zorbaxc18色谱柱(4.6mm×150mm,5μm,美国安捷伦科技公司)流速:0.65ml/min进样量:10μl柱温:35℃对于hplc分析,使用安捷伦(agilent)1100系列(美国安捷伦科技公司)。在240nm处测量λmax,并使用zorbaxc18色谱柱(4.6mm×150mm,5μm,美国安捷伦科技公司)在0.65ml/min、进样量为10μl、柱温为35℃的条件下执行测量。流动相缓冲液使用体积比为2:8的0.1%三氟乙酸(99%)乙腈(无水,99.8%)溶液和去离子水溶液。对于样品处理,将40mg的样品与100ml的缓冲溶剂混合,超声处理10分钟,然后快速搅拌10分钟。使用尼龙注射器式过滤器(孔径为0.2μm)过滤所得溶液,从而测量样品。-sma1和sma2仪器:高效液相色谱(hplc)分析agilent1100系列(美国安捷伦科技公司)紫外线检测器(λmax=230)luna5uc18100a(250×4.6mm,5μm,美国安捷伦科技公司)流速:1ml/min进样量:10μl柱温:30℃对于hplc分析,使用了agilent1100系列(美国安捷伦科技公司)。在230nm处测量λmax,并使用luna5uc18100a(250×4.6mm,5μm,美国安捷伦科技公司)在1ml/min、进样量为10μl、柱温为30℃的条件下执行测量。流动相缓冲液使用体积比为5:5的0.1%三氟乙酸(99%)乙腈(无水,99.8%)溶液和去离子水溶液。对于样品处理,将50mg的样品与100ml的缓冲溶剂混合,超声处理10分钟,然后快速搅拌10分钟。使用尼龙注射器式过滤器(孔径为0.2μm)过滤所得溶液,从而测量样品。2-1:制备实施例1中制备的金属层状氢氧化物的zn、sma1、sma2和aa的量的测量比较了合成工艺之后实施例1-10以及比较实施例1和2中的组分的量。这是确认在合成zlh-sma1和zlh-sma2之后使用vitc的外部离子交换表面处理工艺的表面改性效果的实验。[表11]sma1添加aa添加zn(%)sma1(%)aa(%)实施例10.05135.9586.3438.75实施例20.025137.1403.2340.02实施例30.005138.1440.6441.1实施例40.0025138.2730.3241.21实施例50.0005138.3770.06841.34比较实施例10138.4030.00041.37[表12]sma2添加aa添加zn(%)sma2(%)aa(%)实施例110.05136.4485.0539.15实施例120.025137.4002.5140.01实施例130.005138.1980.5141.06实施例140.0025138.3000.2441.19实施例150.0005138.3820.0541.28比较实施例10138.4030.00041.37当添加sma1或sma2时,调节sma1或sma2的比例,aa相对于总重量的量几乎不改变,并且仅sma1或sma2的量改变。这确认了aa在金属盐层之间没有变化且仅sma1或sma2附着至表面。aa量的轻微变化对应于通过用sma1或sma2代替附着至金属盐表面的aa而引起的数值变化。在xrd上进行sma处理后,没有峰变化,但是通过sma量的测量确认了用sma的改性。[表13][表14]sma2添加aa添加zn(%)sma2(%)aa(%)实施例1610.0522.67963.7511.234实施例1710.02522.93964.1940.617实施例1810.00523.00564.3970.130实施例1910.002523.12964.4880.617实施例2010.000523.13764.5690.039比较实施例31023.15064.6370.000当将sma插入zlh层之间、然后添加vitc、调节vitc的比例时,sma相对于总重量的量几乎不改变,并且仅aa的量改变。这确认了sma在金属盐层之间没有变化,且仅aa结合至表面。sma量的轻微变化对应于通过用sma1或sma2代替附着至金属盐表面的aa而引起的数值变化。在xrd上进行sma处理后,没有峰变化,但是通过sma量的测量确认了用vitc的改性。2-2:制备实施例2中制备的金属层状氢氧化物的zn、sma1、sma2和aa的量的测量[表15][表16]sma2添加aa添加zn(%)sma2(%)aa(%)比较实施例31023.15064.6370实施例310.99950.000522.91364.570.011实施例320.9950.00523.08964.280.113实施例280.50.527.89340.2715.34实施例300.250.7532.34722.8626.47实施例330.0050.99537.9030.50141.01实施例340.00050.999538.3190.04941.21比较实施例10138.403041.37当添加sma1或sma2、sma1或sma2的比例相对于活性成分来调节时,调节aa相对于总重量的量,且也调节sma1或sma2的量。确认了通过将金属盐层之间的sma1或sma2与aa结合在一起来控制量。试验性实施例3.丁达尔效应的测量丁达尔效应基于胶体溶液的光学性能。胶体溶液的颗粒具有1nm-1000nm的粒径,并且显示出该颗粒既不通过范德华力吸引和静电排斥而聚集也不通过范德华力吸引和静电排斥而稳定。胶体颗粒在分散介质中快速移动,称为布朗运动,并且胶体颗粒的尺寸类似于或大于光的波长,因此光无法反射,但发生光散射。因此,丁达尔效应是由于胶体颗粒的光散射而出现的现象。为了观察丁达尔现象,将50mg的各粉末放置于培养皿(petridish)中,然后放置于含有50ml去离子水的小瓶中,然后使用搅拌器均匀分散10分钟。将所有分散体系设定为1000ppm。使溶液静置30分钟,然后使光透射通过激光指示器光束(laserpointerbeam)以观察丁达尔效应。将溶液中的颗粒分散并确认丁达尔效应的情况确定为透射,将颗粒尺寸为1μm以上且发生沉淀的情况确定为沉淀。3-1制备实施例1中制备的金属层状氢氧化物的丁达尔效应的测量实施例1-5和比较实施例1中制备的各金属层状氢氧化物的丁达尔效应在图11-图13中示出,实施例11-15和比较实施例2中制备的各金属层状氢氧化物的丁达尔效应在图14至图16中示出。[表17][表18]当发生丁达尔效应时,可以确认分散性是极好的。在不符合本发明的比较实施例1中,确认产生了沉淀物(图11-图16)。3-2制备实施例2中制备的金属层状氢氧化物的丁达尔效应的测量在上述实验中测量的丁达尔效应在图17至图20中示出。当发生丁达尔效应时,可以确认分散性是极好的。在不符合本发明的比较实施例中,确认产生了沉淀物。[表19][表20]当发生丁达尔效应时,可以确认分散性是极好的。在不符合本发明的比较实施例中,确认产生了沉淀物(图17至图20)。试验性实施例4:颗粒尺寸的评估-仪器:zetasizer纳米zs90分散剂:水分散剂ri:1.330电池:一次性电池运行次数:20温度:25℃使用可购自马尔文帕纳科有限公司(malvernpanalyticalltd.)(uk)的zetasizer、使用电泳光散射测量粉末的颗粒尺寸。将样品分散在去离子水中,然后使用nanozszs90测量颗粒尺寸。4-1:制备实施例1中制备的金属层状氢氧化物的颗粒尺寸的评估通过上述方法测量的实施例1至5以及比较实施例1的颗粒尺寸在下表21中具体示出,且其评估结果在图23中示出。[表21]通过上述方法测量的实施例11至15以及比较实施例1的颗粒尺寸在下表22中具体示出,且其评估结果在图24中示出。[表22]4-2:制备实施例2中制备的金属层状氢氧化物的颗粒尺寸的评估通过上述方法测量的实施例21和24至27以及比较实施例1的颗粒尺寸在下表23中具体示出,且其评估结果在图25中示出。[表23]通过上述方法测量的实施例28和32至34以及比较实施例1的颗粒尺寸在下表24中具体示出,且其评估结果在图26中示出。[表24]试验性实施例5:zeta电位的分析-仪器:zetasizer纳米zs90分散剂:水电池:一次性zeta电池运行次数:30温度:25℃使用可购自malvernpanalyticalltd.(uk)的zetasizer、使用电泳光散射测量分子的表面电位。将样品分散在去离子水中,然后使用nanozszs90测量表面电位。5-1:制备实施例1中制备的金属层状氢氧化物的zeta电位的分析通过上述方法测量的实施例1至5和比较实施例1的zeta电位值在下表25中具体示出,且其具体曲线图在图27中示出。[表25]通过上述方法测量的实施例1至15和比较实施例1的zeta电位值在下表26中具体示出,并且其具体曲线图在图28中示出。[表26]基于通过调节活性成分和表面改性剂的比例而测量的结果,与比较实施例1相比,zeta电位增加,基于此证明了金属层状氢氧化物的表面使用表面改性剂而改性。与比较实施例1或3的使用抗坏血酸相比,zeta电位增加,这是由用sma替代抗坏血酸引起实际改性的现象。5-2:制备实施例2中制备的金属层状氢氧化物的zeta电位的分析通过上述方法测量的实施例21和24至27以及比较实施例6的zeta电位值在下表27中具体示出,且其评估结果在图27中示出。[表27]通过上述方法测量的实施例28和32至34以及比较实施例1的zeta电位值在下表28中具体示出,并且其评估结果在图28中示出。[表28]试验性实施例6:受控释放性的评估针对各粉末的受控释放性,使用了韩国药典的通用测试方法中最常见的桨式(paddle)方法。将300mg的各材料粉末添加到300ml0.08wt%nacl的去离子水溶液(或含有0.08wt%nacl和10%乙醇的溶液)中并分散在其中,然后在25℃、50rpm下搅拌引起测量组分的释放,并且以预定的时间间隔对溶液采样,使用尼龙注射过滤器(0.2μm的孔径)过滤,并使用hplc进行测量。6-1:制备实施例1中制备的金属层状氢氧化物的受控释放性的评估通过上述方法测量的、与实施例1至5以及比较实施例1的受控释放性有关的具体值在下表29中示出,且其具体曲线图在图31中示出。[表29]根据实施例中表面改性剂的比例,抗坏血酸的受控释放性增加。确认了根据表面改性剂的比例,受控释放性在72小时内从最小值约53%增加到最大值约68%,并且还确认了表面改性剂量低于抗坏血酸量,晶体的受控释放性随着存储周期的增加而显著改善。通过上述方法测量的、与实施例11至15以及比较实施例1的受控释放性有关的具体值在下表30中示出,且其具体曲线图在图32中示出。[表30]根据实施例中表面改性剂的比例,抗坏血酸的受控释放性增加。确认了根据表面改性剂的比例,受控释放性在72小时内从最小值约49%增加到最大值约64%,并且还确认了表面改性剂量低于抗坏血酸量,晶体的受控释放性随着存储周期的增加而显著改善。6-2:制备实施例2中制备的金属层状氢氧化物的受控释放性的评估通过上述方法测量的、与实施例21和24至27以及比较实施例1的受控释放性有关的具体值在下表31中示出,且其评估结果在图33中示出。[表31]根据实施例中表面改性剂的比例,抗坏血酸的受控释放性增加。确认了根据表面改性剂的比例,受控释放性在72小时内从最小值约40%增加到最大值约57%,并且还确认了表面改性剂量低于抗坏血酸量,晶体的受控释放性随着存储周期的增加而显著改善。通过上述方法测量的、与实施例28和31至34以及比较实施例1的受控释放性有关的具体值在下表32中示出,且其评估结果在图34中示出。[表32]根据实施例中表面改性剂的比例,抗坏血酸的受控释放性增加。确认了根据表面改性剂的比例,受控释放性在72小时内从最小值约39%增加到最大值约58%,并且还确认了表面改性剂量低于抗坏血酸量,晶体的受控释放性随着存储周期的增加而显著改善。试验性实施例7:使用传统的涂覆剂时,颗粒尺寸和zeta电位的评估[表33]用传统的hpmc、teos和gms涂覆后,通常测得颗粒尺寸为3μm(3000nm)以上,其被评估为大于本发明的颗粒尺寸。zeta电位具有负值,这是针对所使用的涂覆剂所得到的结果。相对小的颗粒尺寸对于皮肤吸收是有利的。试验性实施例8:受控释放性的评估评估了比较实施例4至6和未涂覆的zbs-维生素c(比较实施例1)的受控释放性。其具体结果在图35中示出。如图35所示,用传统涂覆剂涂覆的金属层状氢氧化物复合物的受控释放性在12小时后达到100%。具体地,其证明了与本发明相比,使用传统涂覆剂时的受控释放性非常低。试验性实施例9:实施例1至34和比较实施例1至3的产率和量的确认[表34][表35][表36][表37][表38][表39]工业实用性根据本发明,金属层状氢氧化物复合物包括活性成分和表面改性剂,因此表现出优异的受控释放性。金属层状氢氧化物复合物具有减小的颗粒尺寸,其中活性成分稳定地包含在其中。此外,金属层状氢氧化物复合物可以以如下方式简单地合成:金属层状氢氧化物通过静电吸引结合至具有相反电荷的活性成分,或者调节金属层状氢氧化物的结晶度,从而实现短的处理时间,并避免了专用的制备设施,从而产生了环境友好和经济的效果。另外,本发明提供了一种制备金属层状氢氧化物的方法,该方法避免了由于有机溶剂的使用而引起的后处理、诸如洗涤,并因此在不使用有害材料的情况下是安全的。当前第1页1 2 3
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips