
 商标分类
商标分类  商标转让
商标转让 
一种氟比洛芬的制备方法与流程
 2021-02-02 22:02:14|
2021-02-02 22:02:14| 543|
543| 起点商标网
起点商标网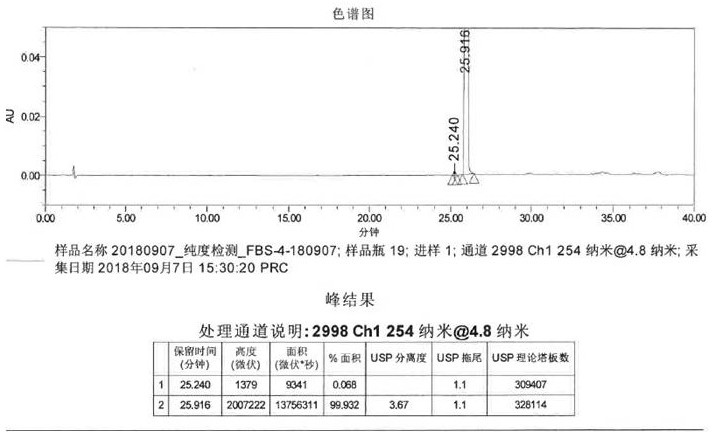
本发明属于药物化合物制备领域,涉及非甾体消炎镇痛药的合成方法,尤其涉及一种氟比洛芬的制备方法。
背景技术:
氟比洛芬又称苯氟布洛芬,化学名为2-(2-氟联苯-4-苯)丙酸,其结构如下所示:
氟比洛芬是一种非甾体抗炎镇痛药。1976年由英国布茨公司研制,已被英国1988年版药典收载。氟比洛芬巴布膏,目前该药已在70多个国家销售,临床上适用于,风湿性关节炎、骨关节炎及弯曲性脊柱炎等,与同类药相比它有计量小、疗效高、起效快、副作用小等优点。截至目前为止,国内原料药主要依赖进口。
该化合物的制备路线最早见于专利us3755427和专利us4266069,其中us3755427采用了七步反应合成氟比洛芬,其中采用ullmann反应高温合成联苯片段,采用高温180~200℃条件脱羧,操作条件苛刻,收率低。专利us4266069采用了六步反应合成氟比洛芬,其中采用致癌物苯作溶剂,重氮化合成联苯片段,合成过程操作困难,毒性大不易于工业放大。
国内氟比洛芬合成工艺路线报道较早,ganglu等人在chinesechemicallettersvol.17,no.4,pp461-464,2006中报道了以2,4-二氟硝基苯为原料经芳香亲核取代,酸水解及加热脱羧,硝基还原,溴化亚铜催化重氮化上溴,钯碳催化四苯硼钠偶联合成氟比洛芬。其中pd/c催化四苯硼钠偶联受铜离子影响收率低。
氟比洛芬合成路线无法避免的问题是氟比洛芬联苯片段的合成,联苯片段合成现有合成技术主要有两种:一种是采用苯作为原料,反应条件苛刻,毒性大;另一种采用苯系物,如卤苯、四苯硼钠、苯硼酸等,需要金属配体催化剂催化反应,成本高。
文献cn106496016a公开了一种制备氟比洛芬的方法,该方法采用3-氟溴苯的格氏试剂与2-溴丙酸钠偶联再酸化得到2-(3-氟苯基)丙酸,2-(3-氟苯基)丙酸溴化得到2-(3-氟-4-溴苯基)丙酸,2-(3-氟-4-溴苯基)丙酸经过金属钯配体催化与苯基硼试剂suzuki偶联反应得到氟比洛芬,在采用金属钯催化偶联进行氟比洛芬合成工艺过程中,首先,金属钯配体催化剂成本高,难以回收,重复利用率极低,且金属钯配体催化反应需要在氮气保护条件下进行,对操作人员及操作设备均有极高的要求,重现性差,其次,在2-(3-氟苯基)丙酸经溴素溴代得到2-(3-氟-4-溴苯基)丙酸的制备过程中,溴素毒性大且对设备要求高,对环境污染严重,最后,在上述制备过程中所用反应物料毒性大,副反应的发生率高,使其严重影响终产品的纯度,难以获得高纯度的终产品,因此,该方法不利于工业生产。
技术实现要素:
本发明提供了一种操作难度更低、重现性高、健康安全,可获得高纯度氟比洛芬的制备方法。
为了实现上述技术目的,本发明开发了一种氟比洛芬新的制备方法,技术方案如下:
本发明公开了一种氟比洛芬的制备方法,该方法即包括氟比洛芬的合成方法,同时还包括氟比洛芬的纯化方法,本方法采用2,4-二氟硝基苯作为起始原料,经亲核取代,水解、加热脱羧、硝基还原,重氮化上溴,钯碳(pd/c)催化苯硼酸偶联反应,合成氟比洛芬,再将氟比洛芬经结晶溶剂纯化,最终获得高纯度的氟比洛芬化合物。
本发明所述的氟比洛芬的制备方法包括将2-(3-氟-4-溴苯基)丙酸与苯硼酸在碱性条件下,加入水体系溶剂,经过pd/c催化的偶联反应,得到氟比洛芬,其中所述的2-(3-氟-4-溴苯基)丙酸与苯硼酸的摩尔用量比为1:1-1:2,2-(3-氟-4-溴苯基)丙酸与pd/c的用量比1:0.1。
本发明所述的氟比洛芬的制备方法,其中所述的2-(3-氟-4-溴苯基)丙酸与苯硼酸的摩尔用量比为1:1-1:1.5,进一步的2-(3-氟-4-溴苯基)丙酸与苯硼酸的摩尔用量比为1:1-1:1.3,在某系些实施例中2-(3-氟-4-溴苯基)丙酸与苯硼酸的摩尔用量比为1:1、1:1.1、1:1.2、1:1.3、1:1.4或者1:1.5。
本发明所述的氟比洛芬的制备方法中2-(3-氟-4-溴苯基)丙酸与pd/c的重量用量比为1:0.05-1:0.5,进一步优选2-(3-氟-4-溴苯基)丙酸与pd/c的重量用量比为1:0.1-1:0.5,在某些实施例中2-(3-氟-4-溴苯基)丙酸与pd/c的重量用量比为1:0.1,1:0.15,1:0.2,1:0.25,1:0.3,1:0.35,1:0.4,1:0.45或者1:0.5。
本发明所述的氟比洛芬的制备方法中,其中所述的催化剂pd/c可重复利用5次以上(含5次),氟比洛芬的产率高于85%。
本发明所述的氟比洛芬的制备方法中,其中所述的催化剂pd/c可重复利用4次以上(含4次),氟比洛芬的产率高于88.95%。
本发明所述的氟比洛芬的制备方法中,其中所述的催化剂pd/c可重复利用4次(含4次)以上,氟比洛芬的产率高于90.97%。
本发明所述氟比洛芬的制备方法中,其中所述的催化剂pd/c可重复利用3次(含3次)以上,氟比洛芬的产率高于91.53%。
本发明所述氟比洛芬的制备方法中,其中所述的催化剂pd/c可重复利用2次(含2次)以上,氟比洛芬的产率高于92.23%。
本发明所述的氟比洛芬的制备方法,其中所述的碱为无机碱,例如氢氧化钠、碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、氢氧化钾等,其中在某些实施例中可选自氢氧化钠、碳酸钠或者碳酸钾,进一步的在某些实施例中所述的碱为碳酸钠。
本发明所述的氟比洛芬的制备方法中,其中所述的碱与2-(3-氟-4-溴苯基)丙酸的摩尔用量比为3.0-4.0,在某些实施例中碱与2-(3-氟-4-溴苯基)丙酸的摩尔用量比为2.1-3.0,进一步的在某系实施例中碱与2-(3-氟-4-溴苯基)丙酸的摩尔用量比2.1、2.2、2.3、2.4、2.5、2.6、2.7、2.8、2.9或者3.0。
本发明所述的氟比洛芬的制备方法,其中所述的水体系溶剂为含有水的溶剂,即可以是单一的水溶剂,也可以水与有机溶剂的混合溶剂,例如水溶剂、醇-水溶剂、四氢呋喃-水溶剂、二氧六环-水溶剂,其中所述的醇可以是甲醇、乙醇或者丙醇,在某些实施例中本发明所述的水体系溶剂是水溶剂、甲醇-水溶剂、四氢呋喃-水溶剂或者二氧六环-水溶剂。
本发明所述的氟比洛芬的制备方法,其中所述的结晶溶剂为醇溶剂和水溶剂,其中醇溶剂可以是甲醇、乙醇或者丙醇,在某系实施例中醇溶剂是甲醇或者乙醇,进一步的在某些实施例中所用的醇溶剂是乙醇溶剂。
本发明所述的氟比洛芬的制备方法中,其中所述的水溶剂,可以选自纯化水、蒸馏水、纯净水等制备常用水。
本发明所述的氟比洛芬的制备方法中,其中所述的醇溶剂,其含量为95%以上的醇溶剂,进一步的为96%以上的醇溶剂,再次为97%以上的醇溶剂,为98%以上的纯溶剂、99%以上的醇溶剂。
本发明所述的氟比洛芬的制备方法中,其中所述的醇溶剂为无水醇溶剂,例如无水甲醇、无水乙醇或者无水丙醇等。
本发明所述的氟比洛芬的制备方法,其步骤包括:
(1)2-(3-氟-4硝基苯基)甲基丙二酸乙酯(fbs-1)制备:采用2,4-二氟硝基苯作为起始原料,在碱性条件下与甲基丙二酸二乙酯亲核取代,生成2-(3-氟-4硝基苯基)甲基丙二酸乙酯;
(2)3-氟-4-硝基-α甲基苯乙酸(fbs-5)制备:在2-(3-氟-4硝基苯基)甲基丙二酸乙酯,加入浓硫酸,溶剂,加热回流脱羧得到3-氟-4-硝基-α甲基苯乙酸,即fbs-5;
(3)4-氨基-3-氟-α甲基苯乙酸(fbs-6)制备:3-氟-4-硝基-α甲基苯乙酸在有机溶剂中,经pd/c氢化得到4-氨基-3-氟-α甲基苯乙酸,即fbs-6;
(4)4-溴-3-氟-α甲基苯乙酸(fbs-7)制备:取fbs-6加入氢溴酸搅拌溶解,冰水浴降温,向体系中滴入亚硝酸钠水溶液;然后将重氮盐体系滴加到cubr和hbr溶液体系中反应,获得得4-溴-3-氟-α甲基苯乙酸;
(5)氟比洛芬的制备:2-(3-氟-4-溴苯基)丙酸与苯硼酸在碱性条件下,加入水体系溶剂,经过pd/c催化的偶联反应,得到氟比洛芬。
本发明所述的氟比洛芬制备方法与现有氟比洛芬的制备方法相比,首先采用pd/c替代金属配体催化剂与苯硼酸联用,避免了使用高费用的金属钯配体催化剂,减低了操作难度,使其更利于工业化生产,其次,催化剂pd/c的可回收多次进行重复利用,且均能获得高产率的氟比洛芬,重复4次回收利用,其产率大于85%,有效降低了生产成本,再次,采用苯硼酸替代其他硼酸试剂,提高反应收率,苯硼酸替代苯,避免致癌物的使用,最后,本发明所述的制备方法,避免了有毒物质、致癌物质及金属物质进行反应,有效降低了环境负荷,更利于绿色化生产,同时,降低了纯化终产品的难度,经本发明所述的纯化工艺纯化氟比洛芬的hplc纯度均达99.5%以上。
说明书附图
图1实施例1纯化后氟比洛芬的hplc谱图;
图2实施例1纯化后氟比洛芬lc-ms谱图;
图3实施例4纯化后氟比洛芬的hplc谱图。
具体实施例
试验材料:
1.试剂类
2、仪器设备类:
磁力搅拌器、三口瓶、布氏漏斗、抽滤瓶、循环水真空泵(shb-b95a型,郑州长城科工贸)、waters液质联用仪(xevog2xs,waters)、waters高效液相色谱仪(e2695,waters)、bruker核磁共振仪(400m)、温度计、恒压滴液漏斗、鼓风干燥箱。
实施例1化合物氟比洛芬的合成
2-(3-氟-4硝基苯基)甲基丙二酸乙酯(fbs-1)的制备
取80ml的n,n-二甲基甲酰胺加入到250ml三口瓶中,加入2.77g氢氧化钠,搅拌不溶,加入11.39g的甲基丙二酸二乙酯搅拌反应,然后加入10.00g2,4-二氟硝基苯,搅拌反应5h,将体系倒至240ml水中淬灭,加入乙酸乙酯萃取(3*100ml),有机相旋干得2-(3-氟-4硝基苯基)甲基丙二酸乙酯19.69g,收率99.75%。
3-氟-4-硝基-α甲基苯乙酸(fbs-5)的制备
取2-(3-氟-4硝基苯基)甲基丙二酸乙酯19.69g,加入至500ml三口瓶中,加入50ml的冰乙酸搅拌溶解,加入13ml的浓硫酸,36ml的水,加热回流24h,加入二氯甲烷萃取(3*60ml)、浓缩、得到油状物3-氟-4-硝基-α甲基苯乙酸10.05g,收率74.83%。
4-氨基-3-氟-α甲基苯乙酸(fbs-6)制备
取3-氟-4-硝基-α甲基苯乙酸油状物10.05g,加入60ml乙醇溶解,加入5%的pd/c(0.50g),氮气气置换三次,通入氢气,在0.1mpa条件下反应6h,过滤,浓缩得到4-氨基-3-氟-α甲基苯乙酸(fbs-6)固体8.10g,收率94.08%。
4-溴-3-氟-α甲基苯乙酸(fbs-7)的制备
取fbs-6固体5.00g,加入20ml氢溴酸(40%)搅拌溶解,冰水浴降温至(0±5℃),向体系中滴入亚硝酸钠水溶液(2.26g/34ml);然后将重氮盐体系滴加到cubr(5.88g)和hbr(10ml)溶液体系中,滴加完毕60℃反应6h,过滤得4-溴-3-氟-α甲基苯乙酸(fbs-7)固体6.10g,收率56.12%。
氟比洛芬的制备
向3000ml三口瓶中加入45g的fbs-7,再加入1800ml纯化水、26.65g苯硼酸、40.54g碳酸钠,4.50gpd/c搅拌升温至回流,保持回流状态反应6h。反应结束后,体系停止加热搅拌降温,至t≤40℃体系过滤,钯碳回收,冰水浴条件下,滤液搅拌降温至5~10℃,浓盐酸调ph=2-3,析出白色固体,过滤,滤饼鼓风干燥箱干燥得白色粉末43.50g,收率97.77%。将白色粉末加入500ml三口瓶中,加入172ml无水乙醇,搅拌溶解,加入0.43g活性炭,搅拌脱色,过滤,滤液转移至500ml三口瓶中,想滤液中滴加172ml纯化水,滴加完毕,控温20±5℃搅拌析晶1h,过滤,滤饼鼓风干燥箱t≤40℃,烘干得41.32g,收率94.98%。
hplc:纯度99.92%;lc-ms:【m-1】(负电离)=243;测熔点:110-111℃。
hnmr(dmso-d6):12.461,1h;7.5547-7.5256,2h;7.5020-7.4562,3h;7.4191-7.3752,1h;7.2513-7.2467,1h;7.2374-7.2167,1h;3.8018-3.7483,1h;1.4203-1.4024,3h。
实施例2重复利用二次pd/c合成化合物氟比洛芬
向1000ml三口瓶中加入15g的fbs-7,再加入600ml纯化水、8.90g苯硼酸、13.50g碳酸钠,1.50gpd/c搅拌升温至回流,保持回流状态反应6h。反应结束后,体系停止加热搅拌降温,至t≤40℃体系过滤,钯碳回收,冰水浴条件下,滤液搅拌降温至5~10℃,浓盐酸调ph=2-3,析出白色固体,过滤,滤饼鼓风干燥箱干燥得白色粉末14.30g,收率96.42%。将白色粉末加入250ml三口瓶中,加入57ml无水乙醇,搅拌溶解,加入0.14g活性炭,搅拌脱色,过滤,滤液转移至250ml三口瓶中,想滤液中滴加57ml纯化水,滴加完毕,控温20±5℃搅拌析晶1h,过滤,滤饼鼓风干燥箱t≤40℃,烘干得13.19g,收率92.23%。
hplc:纯度98.76%。
实施例3重复利用三次pd/c合成化合物氟比洛芬
向1000ml三口瓶中加入10g的fbs-7,再加入400ml纯化水、5.92g苯硼酸、9.01g碳酸钠,0.10gpd/c搅拌升温至回流,保持回流状态反应6h。反应结束后,体系停止加热搅拌降温,至t≤40℃体系过滤,钯碳回收,冰水浴条件下,滤液搅拌降温至5~10℃,浓盐酸调ph=2-3,析出白色固体,过滤,滤饼鼓风干燥箱干燥得白色粉末9.33g,收率94.36%。将白色粉末加入250ml三口瓶中,加入37ml无水乙醇,搅拌溶解,加入0.10g活性炭,搅拌脱色,过滤,滤液转移至250ml三口瓶中,想滤液中滴加37ml纯化水,滴加完毕,控温20±5℃搅拌析晶1h,过滤,滤饼鼓风干燥箱t≤40℃,烘干得8.54g,收率91.53%。
hplc:纯度98.07%。
实施例4:重复利用四次pd/c合成化合物氟比洛芬
向2000ml三口瓶中加入25g的fbs-7,再加入1000ml纯化水、14.80g苯硼酸、22.52g碳酸钠,2.50gpd/c搅拌升温至回流,保持回流状态反应6h。反应结束后,体系停止加热搅拌降温,至t≤40℃体系过滤,钯碳回收,冰水浴条件下,滤液搅拌降温至5~10℃,浓盐酸调ph=2-3,析出白色固体,过滤,滤饼鼓风干燥箱干燥得白色粉末23.15g,收率93.68%。将白色粉末加入250ml三口瓶中,加入92ml无水乙醇,搅拌溶解,加入0.23g活性炭,搅拌脱色,过滤,滤液转移至250ml三口瓶中,想滤液中滴加92ml纯化水,滴加完毕,控温20±5℃搅拌析晶1h,过滤,滤饼鼓风干燥箱t≤40℃,烘干得21.06g,收率90.97%。
hplc:纯度98.14%。
实施例5:重复利用五次pd/c合成化合物氟比洛芬
向2000ml三口瓶中加入20g的fbs-7,再加入800ml纯化水、11.80g苯硼酸、18.01g碳酸钠,10.00gpd/c搅拌升温至回流,保持回流状态反应6h。反应结束后,体系停止加热搅拌降温,至t≤40℃体系过滤,钯碳回收,冰水浴条件下,滤液搅拌降温至5~10℃,浓盐酸调ph=2-3,析出白色固体,过滤,滤饼鼓风干燥箱干燥得白色粉末17.63g,收率90.23%。将白色粉末加入250ml三口瓶中,加入74ml无水乙醇,搅拌溶解,加入0.20g活性炭,搅拌脱色,过滤,滤液转移至250ml三口瓶中,想滤液中滴加74ml纯化水,滴加完毕,控温20±5℃搅拌析晶1h,过滤,滤饼鼓风干燥箱t≤40℃,烘干得17.38g,收率88.95%。
hplc:纯度98.25%。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



