
 商标分类
商标分类  商标转让
商标转让 
一种硝磺草酮杂质、其盐、其互变异构体或其互变异构体的盐及制备方法与流程
 2021-02-02 21:02:02|
2021-02-02 21:02:02| 488|
488| 起点商标网
起点商标网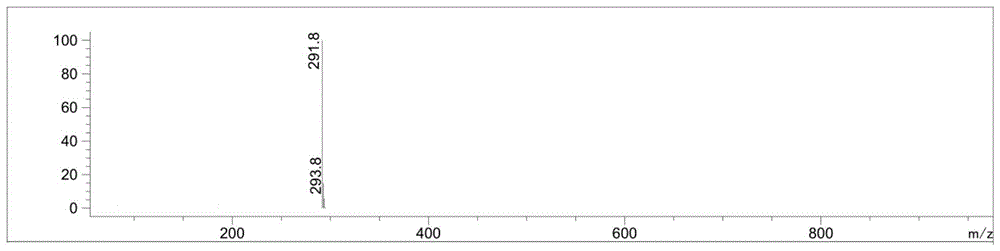
本发明涉及化学合成技术领域,尤其涉及一种硝磺草酮杂质、其盐、其互变异构体或其互变异构体的盐及制备方法。
背景技术:
硝磺草酮是一种能够抑制羟基苯基丙酮酸酯双氧化酶(hppd)的芽前和苗后广谱选择性除草剂,可有效防治主要的阔叶草和一些禾本科杂草。硝磺草酮容易在植物木质部和韧皮部传导,具有触杀作用和特效性。经田间药效试验结果证明,100克/升硝磺草酮悬浮剂对玉米田一年生阔叶杂草和部分禾本科杂草,如苘麻、苋菜、藜、蓼、稗草、马唐等有较好的防治效果,而对铁苋菜和一些禾本科杂草防治效果较差。硝磺草酮的结构式如下。
硝磺草酮在生产和使用过程中会产生一些杂质,这些杂质的存在会使硝磺草酮的外观性状发生变化,并影响硝磺草酮的稳定性,且杂质含量较多,还会导致产品的有效含量降低,影响使用效果,并使硝磺草酮的毒副作用增加,甚至可能对农作物产生负面效果,干扰土壤生物群落结构,影响土壤功能。因此,产品中杂质的检测是保证硝磺草酮除草效果以及减少毒副作用的重要环节。目前,欧盟规定的硝磺草酮杂质主要为4-甲砜基-2-硝基苯甲酸(mnba)、2-氨基-4-甲砜基苯甲酸(amba)两种。为了提高硝磺草酮的用药安全,保证除草效果,有必要充分研究硝磺草酮中含有的杂质。
技术实现要素:
针对现有技术中存在的上述问题,本发明提供一种硝磺草酮杂质、其盐、其互变异构体或其互变异构体的盐及制备方法。
为解决上述技术问题,本发明提供的技术方案是:
一种硝磺草酮杂质化合物1、其盐、其互变异构体或其互变异构体的盐及制备方法:
本发明提供了一种全新的硝磺草酮杂质化合物,为硝磺草酮及其制剂的杂质分析和研究提供了对照品,可用于硝磺草酮原料及其制剂中杂质对照定位、定性或定量使用,为监控生产质量,提升生产工艺提供了研究依据,对于提高硝磺草酮及其制剂的质量可控性具有非常重要的实用价值,有利于提高硝磺草酮的用药安全,减少毒副作用。
本发明还提供了硝磺草酮杂质化合物1、其盐、其互变异构体或其互变异构体的盐的制备方法,包括如下步骤:
溶剂中,硝磺草酮和还原性金属粉在酸性条件下进行还原反应,反应至硝磺草酮的hplc含量小于1%,升温至50-70℃,进行环合反应,得式1所示的化合物、其盐、其互变异构体或其互变异构体的盐。
具体反应过程如下:
本发明提供的硝磺草酮杂质化合物1、其盐、其互变异构体或其互变异构体的盐的制备方法,以硝磺草酮和还原性金属粉为起始物料,原料来源广泛,价格低廉,仅需经过还原反应和环合反应,一锅法即可得到目标产物,且反应过程无需特殊设备,反应条件温和,有利于实现扩大化生产。
优选的,所述酸性条件的ph为1-5,还原反应的温度为20-30℃。
优选的,还原反应中,所述酸性条件用的酸为盐酸、硫酸、磷酸或乙酸中至少一种。
更优选的,还原反应中,所述酸性条件用的酸为盐酸。
上述酸可为市售的化学纯的酸,也可为稀酸溶液,优选的稀酸溶液的浓度为30-40wt%。
优选的,还原反应中,所述溶剂为水、乙腈、四氢呋喃、n,n-二甲基甲酰胺、甲基叔丁基醚或二乙氧基甲烷中至少一种。
优选的,所述还原性金属粉为铁粉、锌粉或镁粉。
优选的,所述硝磺草酮与还原性金属粉的质量比为1:1-3。
更优选的,所述硝磺草酮与还原性金属粉的质量比为1:1.5-2。
优选的,还原反应中,所述溶剂与硝磺草酮的质量比为5-20:1。
更优选的,还原反应中,所述溶剂与硝磺草酮的质量比为5:1。
优选的反应条件,有利于还原反应和环合反应的充分进行,提高原料的转化率,进而提高目标产物的收率和纯度。
优选的,所述硝磺草酮杂质化合物1、其盐、其互变异构体或其互变异构体的盐的制备方法,还包括如下精制步骤:
步骤a,将所得粗品用洗涤溶剂进行洗涤,然后加入碱性溶液,调节ph≥13,进行成盐反应,过滤,得滤液;
步骤b,向所述滤液中加入酸,调节ph为1-3,降温析晶,过滤,得初级精制产品;
步骤c,向所述初级精制产品中加入酸,调节ph为1-3,升温至50-100℃,保温0.5-1h,过滤,将滤液降温析晶,过滤,洗涤,干燥,得产品。
优选的精制步骤,有利于提高目标产物的收率和纯度,精制后的硝磺草酮杂质1的hplc纯度大于99%,收率可达94-98%。
优选的,步骤a中,所述碱性溶液为甲醇钠溶液、甲醇钾溶液、乙醇钠溶液、乙醇钾溶液、氢氧化钠溶液或氢氧化钾溶液。
更优选的,所述述碱性溶液为氢氧化钾溶液。
优选的,所述碱性溶液的浓度为30-35wt%。
优选的,步骤a中,所述洗涤溶剂为水、甲醇、乙醇或异丙醇。
更优选的,步骤a中,所述洗涤溶剂为甲醇。
更优选的,步骤a中,所述洗涤溶剂的加入量为硝磺草酮质量的2-6倍。
进一步优选的,所述洗涤溶剂的加入量为硝磺草酮质量的4倍。
优选的,所述环合反应的温度为60℃。
优选的,所述环合反应的时间为2-4h。
优选的,步骤a中,所述成盐反应的温度为50-90℃,成盐反应的时间为0.5-1h。
更优选的,步骤a中,所述成盐反应的温度为65-75℃。
可选的,步骤b中,所述酸为盐酸、硫酸、乙酸或磷酸。
更优选的,步骤b中,所述酸为盐酸。
优选的,步骤b中,降温析晶的温度为0-30℃,时间为0.5-2h。
更优选的,步骤b中,降温析晶的温度为5-10℃。
优选的,步骤c中,所述降温析晶的温度为0-30℃,时间为0.5-2h。
更优选的,步骤c中,降温析晶的温度为5-10℃。
优选的,步骤c中,升温至60-85℃,保温0.5-1h。
优选的,步骤c中,所述酸为盐酸或硫酸。
更优选的,步骤b中,所述酸为盐酸。
上述酸可为市售的化学纯的酸,也可为稀酸溶液,优选的稀酸溶液的浓度为30-40wt%。
本发明提供的的硝磺草酮杂质化合物1、其盐、其互变异构体或其互变异构体的盐的制备方法,所选试剂和溶剂均为工业领域常用的试剂,廉价易得,操作简单,反应条件温和,制备所得产品的hplc纯度可达99.9%,收率可达94-98%,符合杂质的质量要求,可作为对照品用于硝磺草酮的质量研究,为硝磺草酮的生产和用药安全提供保证。
附图说明
图1为本发明实施例1制备的硝磺草酮杂质化合物1的质谱图;
图2为本发明实施例1制备的硝磺草酮杂质化合物1的液相色谱图;
图3为本发明实施例1制备的硝磺草酮杂质化合物1的红外光谱图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
为了更好的说明本发明,下面通过实施例做进一步的举例说明。
实施例1
一种硝磺草酮杂质化合物1、其盐、其互变异构体或其互变异构体的盐的制备方法:
20℃条件下,向1000ml反应瓶中加入硝磺草酮40g,水800g,开启搅拌,加入质量浓度30wt%的盐酸溶液调节ph至1.1,加入铁粉40g,反应过程中取样液相中控,当硝磺草酮的hplc含量小于1%时,升温至50℃,进行环合反应,反应4h,趁热过滤,所得滤饼用240g异丙醇洗涤,洗涤结束后过滤,将滤饼加入四口瓶中,加入500g水溶解,然后加入质量浓度为30wt%的氢氧化钾溶液,调节ph至13.8,搅拌升温至90℃,保温反应0.5h,趁热过滤,向滤液中加入质量浓度为30wt%的盐酸水溶液,调节ph至1.2,降温至30℃析晶2h,过滤,得粗品。将粗品加入反应瓶中加入水500g,加入质量浓度为30wt%的盐酸水溶液调节ph至1.5,搅拌条件下升温至50℃,保温1h,趁热过滤,向滤液中加入500g冰块,降温至30℃,析晶1h,有黄色产品析出,再次过滤,用冰水洗涤产品,然后于50℃条件下减压干燥,得目标产品33g,液相纯度99.23%,收率95.9%。
1hnmr(43mhz,dmso):δ12.04(1h,s),δ8.41-7.31(3h,m),δ2.80(2h,s),δ2.55(3h,s),δ2.10-1.87(11h,d)。
esi-ms:[m+1]+理论计算值为291.8,试验测定值为291.8,实际值与理论值相符。质谱谱图如图1所示,液相谱图如图2所示。
制备的目标产品的红外光谱图如图3所示。
实施例2
一种硝磺草酮杂质化合物1、其盐、其互变异构体或其互变异构体的盐的制备方法:
30℃条件下,向1000ml反应瓶中加入硝磺草酮40g,二乙氧基甲烷400g,开启搅拌,加入质量浓度30wt%的硫酸溶液调节ph至4.8,加入锌粉120g,反应过程中取样液相中控,当硝磺草酮的hplc含量小于1%时,升温至70℃,进行环合反应,反应2h,趁热过滤,所得滤饼用90g乙醇洗涤,洗涤结束后过滤,将滤饼加入四口瓶中,加入水100g溶解,然后加入质量浓度为30wt%的氢氧化钠溶液,调节ph为14.1,搅拌升温至50℃,保温反应1h,趁热过滤,向滤液中加入质量浓度为30wt%的盐酸水溶液,调节ph至2.8,降温至0℃析晶0.5h,过滤,得粗品。将粗品加入反应瓶中加入水200g,加入质量浓度为30wt%的盐酸水溶液调节ph至2.7,搅拌条件下升温至100℃,保温0.5h,趁热过滤,向滤液中加入200g冰块,降温至0℃,析晶0.5h,有黄色产品析出,再次过滤,用冰水洗涤产品,然后于50℃条件下减压干燥,得目标产品32.5g,液相纯度99.55%,收率94.6%。
1hnmr(43mhz,dmso):δ12.04(1h,s),δ8.41-7.31(3h,m),δ2.80(2h,s),δ2.55(3h,s),δ2.10-1.87(11h,d)。
esi-ms:[m+1]+理论计算值为291.8,试验测定值为291.8,实际值与理论值相符。
实施例3
一种硝磺草酮杂质化合物1、其盐、其互变异构体或其互变异构体的盐的制备方法:
25℃条件下,向1000ml反应瓶中加入硝磺草酮40g,甲基叔丁基醚200g,开启搅拌,加入质量浓度30wt%的盐酸溶液调节ph至2.5,加入铁粉60g,反应过程中取样液相中控,当硝磺草酮的hplc含量小于1%时,升温至60℃,进行环合反应,反应3h,趁热过滤,所得滤饼用80g甲醇洗涤,洗涤结束后过滤,将滤饼加入四口瓶中,加入水350g溶解,然后加入质量浓度为35wt%的甲醇钠溶液,调节ph至13.8,搅拌升温至70℃,保温反应0.5h,趁热过滤,向滤液中加入质量浓度为30wt%的盐酸水溶液,调节ph至1.8,降温至5℃析晶1h,过滤,得粗品。将粗品加入反应瓶中加入水200g,加入质量浓度为30wt%的盐酸水溶液调节ph至1.2,搅拌条件下升温至75℃,保温1h,趁热过滤,向滤液中加入500g冰块,降温至10℃,析晶1h,有黄色产品析出,再次过滤,用冰水洗涤产品,然后于50℃条件下减压干燥,得目标产品33.8g,液相纯度99.90%,收率98.2%。
1hnmr(43mhz,dmso):δ12.04(1h,s),δ8.41-7.31(3h,m),δ2.80(2h,s),δ2.55(3h,s),δ2.10-1.87(11h,d)。
esi-ms:[m+1]+理论计算值为291.8,试验测定值为291.8,实际值与理论值相符。
将本发明实施例1中的反应条件,如温度、时间、溶剂等替换为本发明说明书中限定的其他条件,均可达到与实施例1基本相当的技术效果。
对比例1
本对比例提供一种硝磺草酮杂质化合物1的合成方法:
向四口瓶中加入硝磺草酮50g,加水400ml,开启搅拌,50℃下滴加质量浓度为40wt%的氢氧化钾溶液至料液溶清。溶清后将料液投入高压釜内,投入雷尼镍5g,密闭高压釜,依次用氮气、氢气置换高压釜内气体。然后升温至50-60℃,缓慢通入氢气,维持压力0.3-0.4mpa,温度50-60℃,至压力不再下降为反应终点。用氮气将料液经过滤器压出,压出后的料液在氮气保护下,用质量浓度为30wt%的硫酸溶液酸化至中性,然后加热至80-90℃,同时加入5g三氯化铁做催化剂,保温反应,反应过程中取样hplc检测进行中控,至中间体含量小于1%反应结束,然后降至室温,过滤,水洗,得硝磺草酮杂质化合物1,hplc含量46%,硝磺草酮原料剩余36%,主要未知杂质6.95%。具体反应过程如下:
反应过程中控中间体含量的检测方法为:色谱柱:c18柱(4.6×250mm,5μm);
流动相:流动相a:流动相b=40:60;流动相a为甲醇;流动相b为0.02mol/lkh2po4水溶液(h3po4调ph为3);
检测波长:220nm;
流速:0.8ml/min。
对比例2
本对比例提供一种硝磺草酮杂质化合物1的合成方法:
取4.87g三光气溶于10.94g四氢呋喃中,溶清后待用。向四口瓶中加入邻氨基对甲砜基苯甲酸10.11g和四氢呋喃10.94g,升温至50-60℃,滴加三光气的四氢呋喃溶液。hplc中控原料含量<0.5%后过滤,将滤饼溶于四氢呋喃中,得中间体的四氢呋喃溶液,待用。
向另一四口瓶中,加入1.09g环己二酮,5.00g四氢呋喃和1.30gnah,升温至90℃,缓慢滴加中间体的四氢呋喃溶液,滴加完成后保温中控,至中间体含量小于1%反应结束,然后降至室温,过滤,水洗,得硝磺草酮杂质化合物1,hplc含量78.0%,中间体含量9.88%,主要未知杂质含量10.10%。具体反应过程如下:
反应过程中控中间体含量的检测方法为:色谱柱:c18柱(4.6×250mm,5μm);
流动相:流动相a:流动相b=40:60;流动相a为甲醇;流动相b为0.02mol/lkh2po4水溶液(h3po4调ph为3);
检测波长:220nm;
流速:0.8ml/min。
对比例3
本对比例提供一种硝磺草酮杂质化合物1的合成方法:
向四口瓶中加入邻氨基对甲砜基苯甲醛5.0g、水20.0g,质量浓度为32%的盐酸溶液2.0g,搅拌均匀后,升温至50℃,加入环己二酮3.0g,三氯化铁1.0g,维持温度50℃,保温中控,反应6h后,降至室温,过滤,水洗,得硝磺草酮杂质化合物1,目标产物液相全面积含量25.36%,原料邻氨基对甲砜基苯甲醛剩余46%,环己二酮剩余10.1%,主要未知杂质含量为12.80%。具体反应过程如下:
实施例1-3以及对比例1-3中硝磺草酮杂质化合物1及其中杂质的含量检测的hplc方法为:
色谱柱:c18柱(4.6×250mm,5μm);
流动相:流动相a:流动相b=40:60;流动相a为甲醇;流动相b为0.02mol/lkh2po4水溶液(h3po4调ph为3);
检测波长:220nm;
流速:0.8ml/min。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换或改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



