
 商标分类
商标分类  商标转让
商标转让 
FAPα特异性识别的具有肿瘤诊断治疗功能的亚甲蓝衍生物及其制备方法和应用与流程
 2021-02-02 21:02:19|
2021-02-02 21:02:19| 486|
486| 起点商标网
起点商标网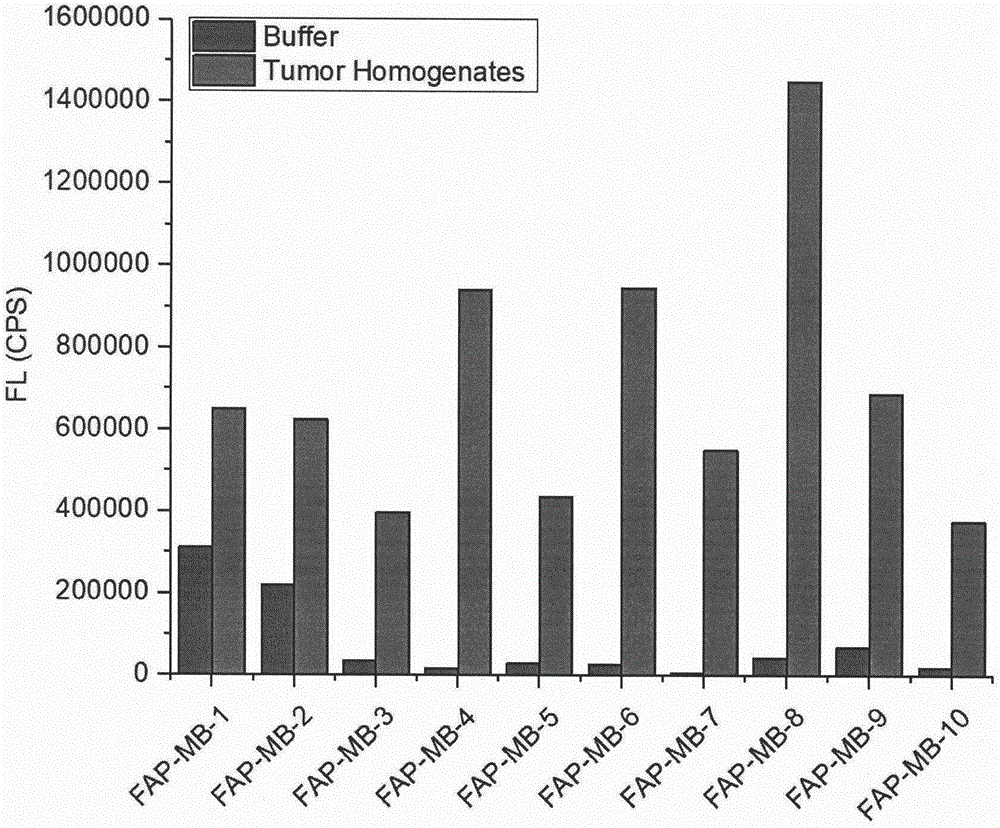
发明书摘要
本发明涉及通式i所示的fapα特异性识别的亚甲蓝衍生物及它们药学上可接受的盐、水合物或前药及其制备方法。其中基团x和r具有在说明书中给出的含义。本发明还涉及通式i的化合物及其药学上可接受的盐、水合物或前药在乳腺癌等fapα高表达实体瘤诊断和光动力治疗中的用途,特别是在制备诊断和治疗癌症的药物中的用途。
fapα特异性识别的具有肿瘤诊断治疗功能的亚甲蓝衍生物及其制备方法和应用
本发明涉及新颖的含亚甲蓝结构的fapα特异性识别的化合物及其药学上可接受的盐、水合物或其前药,它们的制备方法以及含有所述化合物的药物组合物。本发明还涉及该类化合物肿瘤诊断作用及肿瘤部位较强的光动力治疗作用,并且还涉及该类化合物及其药学上可接受的盐、水合物、溶剂化物或其前药在制备诊断和治疗高表达fapα的实体瘤的药物中的用途,特别是在制备诊断和治疗高表达fapα的实体瘤的药物中的用途。
背景技术:
癌症全球范围内的医学难题,是一类威胁人类生命健康的重大难治性疾病,其死亡率甚至超过心脑血管类疾病,居于所有疾病首位。目前肿瘤诊断仍存在灵敏度差等缺点,常用肿瘤治疗药物也存在靶向性差或特性差的问题,同时肿瘤治疗也缺乏实时有效的治疗检测手段。因此,建立便利、灵敏和准确的肿瘤诊断及治疗方法是必要的。
亚甲蓝(methyleneblue,mb)作为一种有机近红外荧光染料,其用于肿瘤荧光诊断已受广泛关注。另外,亚甲蓝还是一种吩噻嗪类光敏剂,已被临床用于光动力治疗。但是亚甲蓝对肿瘤亲和力差及肿瘤组织潴留时间短等缺点限制了其临床应用。
成纤维激活蛋白(fibroblastactivationprotein,fap)是由活化的肿瘤相关成纤维细胞表达的一种膜型丝氨酸蛋白酶,在肿瘤-宿主界面基质的降解和重建中发挥着重要作用,参与肿瘤的生长、浸润和转移,而在正常组织中几乎不表达。fap包括fapα和fapβ两种亚基,其中,fapα具有胶原酶、二肽激酶和肽链内切酶活性。由于其可特异性识别并水解n-末端封端的甘氨酰脯氨酸与其他氨基酸或小分子氨基形成的肽键,fapα已逐渐发展为一种有效的抗肿瘤靶标。
根据文献报道和本发明前期工作基础,发明人发现当亚甲蓝吩噻嗪母核中的氮原子有其他取代基时,荧光衰弱,同时丧失光动力活性。因此,本发明人基于亚甲蓝为诊断和光动力治疗的药物分子,对其母核氮原子进行肿瘤fapα靶向断裂修饰,所设计合成了一系列靶向肿瘤基质的诊断与治疗的新型小分子。该类分子本身没有荧光性质和光动力活性,进入生物体内靶向肿瘤部位经fapα剪切裸露出氨基后自断裂形成亚甲蓝荧光恢复并发挥光动力活性。体外活性筛选表明该类化合物具有较好的肿瘤靶向性和诊疗效果。
技术实现要素:
本发明的主要目的在于提供可作为特异性靶向肿瘤fapα的诊断与光动力治疗式i结构的新型化合物,特别地,本发明还涉及该类化合物的制备方法和包括上述化合物及其异构体的组合物,还涉及该类化合物及其药学上可接受的盐、水合物、溶剂化物或其前药在制备诊断与治疗fapα高表达肿瘤的用途。
本发明涉及通式i的化合物、光学异构体及其药学上可接受的盐、水合物或前药,
其中,
x任选自o,nh和s;
y为亚甲基或
r为氢、
r1和r2分别独立地选自c1-c10烷基、c3-c7环烷基、c3-c7杂环烷基、c2-c10烯基、c2-c10炔基、c6-c10芳基、5-10元杂芳基、稠环芳基、c6-c10杂环基、任选被一个或多个羟基、氨基、卤素、氰基、硝基、磺酰基、芳环、芳杂环和稠环芳基取代的c1-c6烷基;
本发明优选涉及通式i的化合物、光学异构体及其药学上可接受的盐、水合物或前药,其中,
其中,
x任选自o和nh;
y为亚甲基或
r为氢、
r1和r2分别独立地选自c1-c6烷基、c3-c6环烷基、c3-c5环烷基、c3-c7杂环烷基、c2-c8烯基、c2-c4炔基、c6-c10芳基、5-10元杂芳基、稠环芳基、c6-c10杂环基、任选被一个或多个羟基、氨基、卤素、氰基、硝基、芳环、芳杂环或稠环芳基取代的c1-c6烷基;
本发明特别优选涉及通式i的化合物、光学异构体及其药学上可接受的盐、水合物或前药,其中,
x任选自o和nh;
y为亚甲基或
r为氢、
r1和r2分别独立地选自c1-c6烷基、c3-c6环烷基、c2-c8烯基、c2-c4炔基、c6-c10芳基、5-10元杂芳基、稠环芳基、任选被一个或多个羟基、氨基、卤素、氰基、硝基、芳环、芳杂环或稠环芳基取代的c1-c6烷基;
本发明还特别优选涉及通式i的化合物、光学异构体及其药学上可接受的盐、水合物或前药,其中,
x任选自o;
y为
r为氢、
r1和r2分别独立地选自c1-c6烷基、c3-c6环烷基、c2-c6烯基、c2-c4炔基、苯基、吡啶、吲哚、噻吩、喹啉、任选被一个或多个氨基、卤素、苯环、吡啶环、喹啉、吲哚环取代的c1-c6烷基;
本发明通式i的化合物、光学异构体及其药学上可接受的盐、水合物或前药优选以下化合物但这些化合物并不意味着对本发明的任何限制:
[4-(甘氨酰脯氨酰胺基)苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-(n-乙酰基甘氨酰脯氨酰胺基)苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-(n-丙酰基甘氨酰脯氨酰胺基)苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-[n-(2-甲基丙烯酰基)甘氨酰脯氨酰胺基]苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-[n-(2-二甲氨基乙酰基)甘氨酰脯氨酰胺基]苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-(苯丙氨酰甘氨酰脯氨酰胺基)苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-[n-(1h-吲哚-2-丙酰)甘氨酰脯氨酰胺基]苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-[n-(叔丁氧羰基)甘氨酰脯氨酰胺基]苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-[n-(苄氧羰基)甘氨酰脯氨酰胺基]苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
[4-[n-[1-(吡啶-4-基)甲氧羰基]甘氨酰脯氨酰胺基]苄基]-3,7-二(二甲氨基)-10h-吩噻嗪-10-羧酸酯;
一种药用组合物,包含权利要求1-5中任何一项化合物、光学异构体及其药学上可接受的盐、溶剂化物或前药作为活性成分以及药学上可接受的赋形剂。
而且,按照本发明所属领域的一些通常方法,本发明的通式i的化合物及光学异构体可以与酸生成它的药学上可接受的盐。酸可以包括无机酸或有机酸,与下列酸形成的盐是特别优选的:盐酸、氢溴酸、硫酸、磷酸、甲磺酸、乙磺酸、甲苯磺酸、苯磺酸、萘二磺酸、乙酸、丙酸、乳酸、三氟乙酸、马来酸、柠檬酸、富马酸、洒石酸、苯磺酸、苯甲酸或对甲苯磺酸等。
本发明所用的术语“光学异构体”包括r型异构体和s型异构体。本发明通式i的化合物,抗肿瘤活性是用其外消旋体测试所得。
本发明还涉及通式i的化合物及光学异构体具有强的fapα靶向肿瘤的诊断和治疗作用,并且还涉及该类化合物及其药学上可接受的盐、水合物在制备诊断及治疗fapα高表达肿瘤的用途。
下面合成路线1-4描述了本发明的通式i化合物的制备,所有原料都是通过合成路线中描述的方法、通过有机化学领域普通技术人员熟知的方法制备或者可商购。本发明的全部最终化合物都是通过合成路线中描述的方法或通过与其类似方法制备的,这些方法是有机化学领域普通技术人员熟知的。合成路线中应用的全部可变因数如下文定义或权利要求中定义。
按照本发明的式i衍生物,均可按照合成路线1的方法由亚甲蓝经还原、酰胺化、酰化、水解和酰化得到。
附图说明
图1表示不同目标化合物与高表达fapα肿瘤组织匀浆孵育后荧光强度变化。
具体实施方式
实施例旨在阐述而不是限制本发明的范围。化合物的核磁共振氢谱用brukerarx-300测定,质谱用agilentl100lc/msd测定;所用试剂均为分析纯或化学纯。
制备通法:
步骤a3,7-二甲氨基-10h-吩噻嗪-10-甲酰氯蓝(iii)
室温下,亚甲蓝溶于10ml水和30ml的二氯甲烷的混合溶剂,加入碳酸钠,于40℃搅拌5min,连二亚硫酸钠溶于15ml水滴加入上述反应液,40℃搅拌1h,溶液由蓝色变为棕黄色,显示还原反应完成,tlc监测反应毕,得到中间体ii,未经纯化,将反应液冷却至0℃冰浴,缓慢向其滴加三光气的二氯甲烷溶液10ml,冰浴搅拌下,滴加至上述反应液,维持冰浴条件,继续搅拌2h,反应毕,将反应液倒入100ml冰水中淬灭反应,硅藻土抽滤,二氯甲烷洗涤滤饼,收集滤液,将滤液置于分液漏斗中,dcm(50ml*3)萃取,饱和食盐水(60*3)洗,无水硫酸钠干燥,浓缩,柱层析纯化,得到白色固体。
步骤bn-叔丁氧羰基甘氨酰脯氨酸衍生物(iv)
室温下,将boc-甘氨酸-l-脯氨酸溶于dmf10ml中,加入hatu和dipea,将反应液置于冰浴条件下,0℃下,向反应液缓慢滴加相应的胺的dmf溶液5ml,维持冰浴搅拌30min,室温条件继续搅拌4h,溶液一直呈棕褐色,反应毕,旋转蒸除溶剂,残留物加入20ml水,二氯甲烷(50ml*3)萃取,饱和食盐水(50ml*3)洗,无水硫酸钠干燥,浓缩,柱层析纯化,得到黄色固体。
步骤c中间体v的合成
室温下,将化合物iii溶于10ml二氯甲烷中,加入dmap和na2co3,将反应液置于0℃冰浴条件下10min,将化合物iv溶于5ml二氯甲烷中,冰浴搅拌下,滴加至上述反应液,室温反应过夜,tlc监测反应毕。反应液抽滤,滤除不溶物,滤液蒸干,柱层析纯化,得到蓝色固体。
步骤d中间体vi的合成
室温下,将化合物v溶于dcm中,冰浴下滴加tfa,加毕,室温反应2h,tlc监测反应完全,用5%naoh溶液调节ph=12,分离有机层,饱和食盐水(50ml*3)洗,无水硫酸钠干燥,蒸干得蓝色固体。
步骤e目标产物i的合成
方法一:室温下,化合物vi溶于二氯甲烷10ml中,加入相应酸酐和dmap,室温继续搅拌过夜,tlc监测反应毕,反应液浓缩,柱层析纯化,得到淡蓝色固体。
方法二:室温下,化合物vi溶于二氯甲烷10ml中,加入相应的酰氯(或
按照制备通法,分别制备实施例1-化合物(见表一)。
表一:
对按照本发明的上式i化合物进行了特异性fapα靶向及荧光恢复(诊断)实验筛选。
(1)组织匀浆缓冲液(50mmtris-hcl,100mmnacl)配制:称取3.03gtris碱,2.92gnaci,用ddh2o定容至500ml容量瓶,再用盐酸调ph至7.4,4℃低温保存。
(2)fap-mb-1~10前药储备液配制:精确称取适量的前药,溶于适量的dmso中,配制成浓度为1mm的前药储备液,4℃低温保存。
待高表达fapα肿瘤长到合适大小后(100-150mm3(tumorvolume)),处死荷瘤小鼠,解剖出肿瘤组织,置于培养皿中,冰浴条件下将组织尽可能剪碎,倒入匀浆器中,按每200mg组织加入800ul组织匀浆缓冲液,冰浴充分研磨直至无肉眼可见组织块,3500rpm低温离心15min,离心后得上层组织匀浆液,每个1.5mlep管中加入400ul肿瘤匀浆液,同时阴性对照组每管加入400ul空白缓冲液,每管分别加入3ul的fap-mb-1~10前药储备液,将ep管封口放入恒温摇床中37℃、200rpm孵育12h后,各加入1ml乙腈沉淀蛋白,涡旋至上下两层混匀,10000rpm离心15min,离心后取上清液,0.45um有机滤膜滤除不溶物,所得溶液各补加1ml乙腈,避光保存,待进行荧光光谱扫描实验。
荧光光谱扫描实验:
固定激发波长λex=633nm;发射波长λem=650~720nm;狭缝宽度为5对每个前药孵育的肿瘤匀浆液和空白缓冲液进行扫描。记录每个化合物的荧光曲线和荧光数值进行比较。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



