
 商标分类
商标分类  商标转让
商标转让 
一种长链星型支化剂及高流动高结晶长碳链星型聚酰胺的制备方法与流程
 2021-02-02 20:02:12|
2021-02-02 20:02:12| 471|
471| 起点商标网
起点商标网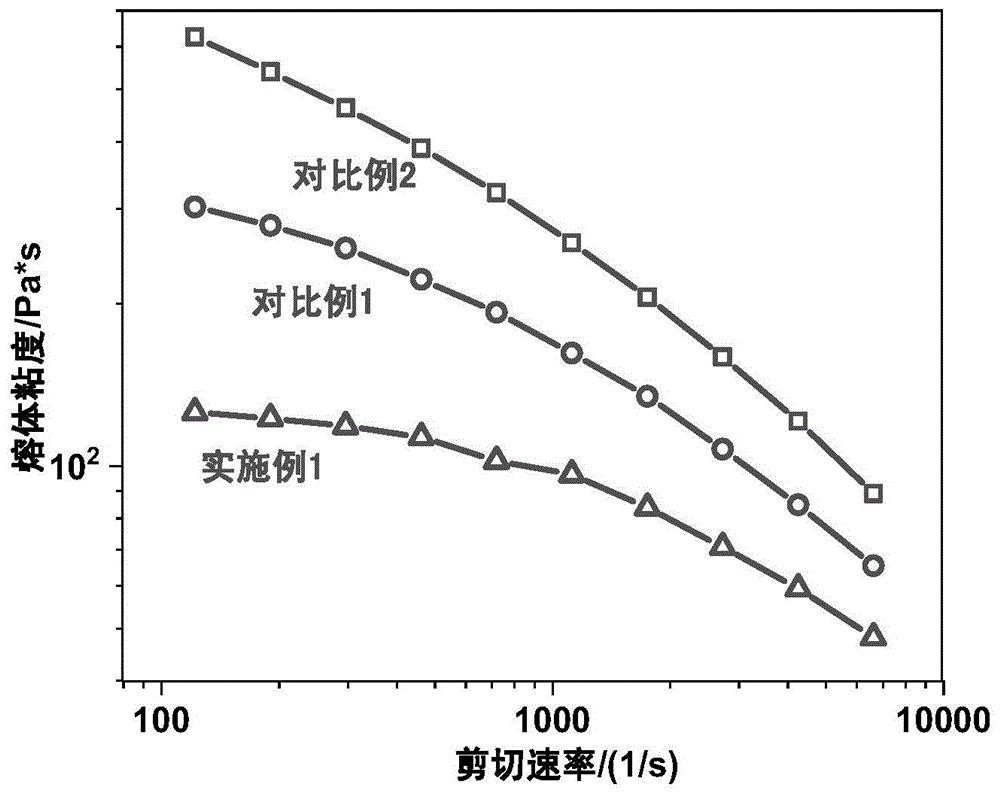
本发明涉及星型聚酰胺材料制备领域,特别是涉及一种长链星型支化剂及高流动高结晶长碳链星型聚酰胺的制备方法。
背景技术:
星型聚酰胺是一类不少于三条链(臂),且支链都以化学键的形式连接于核上的支化聚酰胺。相比于传统的线型聚酰胺,星型聚酰胺具有一定的三维网状结构,由此减少了线型聚酰胺链段之间的缠绕,熔融状态下剪切变稀性更为明显,可以有效地降低聚酰胺加工时熔体的粘度,增加其流动性,通常可以作为改性材料来增加玻纤增强复合材料的流动性、当作为导热材料时可以加入更多的阻燃和绝缘助剂和用作注塑磁体以及其他外形复杂、薄壁的制件。
目前,现有技术中在利用星型聚酰胺制备高流动性制件的同时虽然保证了一定的力学强度,但分析总结星型聚酰胺的力学测试指标可以发现:1)在聚合物中引入一定程度的支化结构后,会导致材料的韧性和抗冲强度均出现不同程度的下降,如trouillet-fonti等使用三官能度的醚胺作为支化剂,配合一元/二元羧酸作为分子量调节剂制备的星型pa66产品缺口冲击强度下降了39%-57%,断裂伸长率从30%降低至1.7-2%;nijenhuis的专利中加入1.4%的星型支化剂2,4,6-三(氨基己酸基)-1,3,5-三嗪(tact)后pa6的断裂伸长率由55%降低至30%;2)此外,星型聚酰胺自身无链缠绕、结晶度低,结晶速度显著低于线性聚酰胺,结晶性能的下降带来材料注塑成型周期的延长,降低了生产效率。
技术实现要素:
鉴于以上所述现有技术的缺点,本发明的目的在于提供一种长链星型支化剂及高流动高结晶长碳链星型聚酰胺的制备方法,用于解决现有技术中的问题。制备获得的长碳链星型聚酰胺拥有高的流动性和良好的抗冲击性能,同时兼具高结晶性能,可以有效缩短成型周期。
为实现上述目的及其他相关目的,本发明是通过以下技术方案获得的。
本发明一方面提供一种长链星型支化剂的制备方法,所述长链星型支化剂的制备方法包括如下步骤:
1)将环己烷二甲酰氯与长碳链二元胺、第一缚酸剂通过界面聚合反应,制备获得低分子量聚合物;
2)将环己烷烷基酰氯与步骤1)所述的低分子量聚合物进一步反应制备获得长链星型支化剂;其中,所述环己烷烷基酰氯的官能度≥3。
在本发明的一些实施方式中,所述步骤1)中,所述环己烷二甲酰氯的制备方法包括将环己烷二甲酸进行酰氯化反应制备获得环己烷二甲酰氯;优选地,所述环己烷二甲酰氯的制备方法包括向环己烷二甲酸中加入第一溶剂和第一催化剂反应后,提纯、蒸馏、干燥制备获得。
在本发明的一些实施方式中,所述步骤1)中,所述长碳链二元胺选自壬二胺、癸二胺、十一碳二元胺、十二碳二元胺中的一种或多种的组合。
在本发明的一些实施方式中,所述步骤1)中,所述第一缚酸剂选自四丁基氢氧化胺,三乙胺,n,n-二异丙基乙胺,4-二甲氨基吡啶,碳酸钠,碳酸氢钠,尿素中的一种或多种的组合。
在本发明的一些实施方式中,所述步骤2)中,所述环己烷烷基酰氯的制备方法包括将环己烷烷基酸进行酰氯化反应制备获得环己烷烷基酰氯;优选地,所述环己烷烷基酰氯的制备方法包括向环己烷烷基酸中加入第二溶剂和第二催化剂反应后,提纯、蒸馏、干燥制备获得;其中,所述环己烷烷基酸的官能度≥3。
在本发明的一些实施方式中,所述环己烷二甲酰氯选自1,4-环己烷二甲酰氯。
在本发明的一些实施方式中,所述环己烷烷基酰氯为中心对称结构;优选的,所述环己烷烷基酰氯中酰氯取代基团在环己烷的1,2,4,5位或者1,3,5位;更优选的,所述环己烷烷基酰氯选自1,2,4,5-环己烷四甲酰氯、1,3,5-环己烷三甲酰氯、1,3,5-环己烷三乙酰氯中的一种或多种的组合。
在本发明的一些实施方式中,所述低分子量聚合物的分子量为200~2000。
在本发明的一些实施方式中,所述长链星型支化剂的分子量为1400~10000。
在本发明的一些实施方式中,所述环己烷二甲酸选自1,4-环己烷二甲酸。
在本发明的一些实施方式中,所述环己烷烷基酸为中心对称结构;优选的,所述环己烷烷基酸中酸取代基团在环己烷的1,2,4,5位或者1,3,5位;更优选的,所述环己烷烷基酸选自1,2,4,5-环己烷四甲酸、1,3,5-环己烷三甲酸、1,3,5-环己烷三乙酸中的一种或多种的组合。
在本发明的一些实施方式中,所述第一溶剂选自三光气的甲苯溶液。
在本发明的一些实施方式中,所述第一催化剂选自n,n-二甲基甲酰胺、吡啶、三乙胺、咪唑中的一种或多种的组合。
在本发明的一些实施方式中,所述第二溶剂选自三光气的甲苯溶液。
在本发明的一些实施方式中,所述第二催化剂选自n,n-二甲基甲酰胺、吡啶、三乙胺、咪唑中的一种或多种的组合。
在本发明的一些实施方式中,所述长碳链二元胺和环己烷烷基酸的摩尔比为0.5~4:1。
在本发明的一些实施方式中,所述第一缚酸剂和环己烷烷基酸的摩尔比为0.8~10:1。
在本发明的一些实施方式中,所述第一催化剂和环己烷烷基酸的质量比为1:1~5。
在本发明的一些实施方式中,所述第二催化剂和环己烷烷基酸的质量比为1:1~5。
本发明另一方面提供一种长链星型支化剂的制备方法,所述长链星型支化剂的制备方法包括:将本发明所述的环己烷烷基酰氯、长碳链内酰胺与第二缚酸剂反应制备获得长链星型支化剂;其中,所述环己烷烷基酸的官能度≥3。
在本发明的一些实施方式中,所述环己烷烷基酰氯的制备方法包括将环己烷烷基酸进行酰氯化反应制备获得环己烷烷基酰氯;其中,所述环己烷烷基酸的官能度≥3。
在本发明的一些实施方式中,所述的长碳链内酰胺选自十一碳内酰胺和/或十二碳内酰胺。
在本发明的一些实施方式中,所述第二缚酸剂选自四丁基氢氧化胺,三乙胺,n,n-二异丙基乙胺,4-二甲氨基吡啶,碳酸钠,碳酸氢钠,尿素中的一种或多种的组合。
在本发明的一些实施方式中,所述环己烷烷基酰氯为中心对称结构;优选的,所述环己烷烷基酰氯中酰氯取代基团在环己烷的1,2,4,5位或者1,3,5位;更优选的,所述环己烷烷基酰氯选自1,2,4,5-环己烷四甲酰氯、1,3,5-环己烷三甲酰氯、1,3,5-环己烷三乙酰氯中的一种或多种的组合。
在本发明的一些实施方式中,所述长碳链内酰胺和环己烷烷基酸的摩尔比为0.5~4:1。
在本发明的一些实施方式中,所述第二缚酸剂和环己烷烷基酸的摩尔比为0.8~10:1。
在本发明的一些实施方式中,所述环己烷烷基酸为中心对称结构;优选的,所述环己烷烷基酸中酸取代基团在环己烷的1,2,4,5位或者1,3,5位;更优选的,所述环己烷烷基酸选自1,2,4,5-环己烷四甲酸、1,3,5-环己烷三甲酸、1,3,5-环己烷三乙酸中的一种或多种的组合。
本发明另一方面提供一种长链星型支化剂,由本发明所述的长链星型支化剂的制备方法制备获得。
本发明另一方面提供一种长碳链星型聚酰胺,所述长碳链星型聚酰胺的制备原料包括长碳链尼龙盐、本发明所述的长链星型支化剂、助剂和水。
在本发明的一些实施方式中,所述助剂包括第三催化剂、消泡剂、抗氧剂、分子量调节剂。
在本发明的一些实施方式中,所述长碳链星型聚酰胺按其重量份计,包括如下组分:
在本发明的一些实施方式中,所述长碳链尼龙盐选自癸二酰己二胺盐、癸二酰癸二胺盐、十二烷二酰己二胺盐、十二烷二酰癸二胺盐、十二烷二酰十二烷二胺盐中的一种或多种的组合。
在本发明的一些实施方式中,所述第三催化剂选自次磷酸水溶液,次亚磷酸钠、次亚磷酸钾、次亚磷酸钙中的一种或多种的组合。
在本发明的一些实施方式中,所述消泡剂选自聚乙二醇、聚乙二醇-丙二醇共聚物、聚二甲基硅氧烷中的一种或多种的组合。
在本发明的一些实施方式中,所述抗氧剂选自抗氧剂1098、抗氧剂168和抗氧剂1010中的一种或多种的组合。
在本发明的一些实施方式中,所述分子量调节剂选自苯甲酸、醋酸、长链二元酸和长链二元胺中的一种或多种的组合。
本发明另一方面提供本发明所述的长碳链星型聚酰胺的制备方法,包括将长链星型支化剂与长碳链尼龙盐以及助剂混合后进行聚合反应制备获得。
附图说明
图1为本发明实施例1、对比例1和对比例2的熔体粘度示意图。
具体实施方式
本发明的申请人经过实验发现,通过合成一种环己烷二甲酸和长链二元胺作为内臂,官能度≥3的环己烷烷基酸(例如环己烷烷基酰氯为中心对称结构,例如所述环己烷烷基酰氯中酰氯取代基团在环己烷的1,2,4,5位或者1,3,5位,更例如环己烷四甲酸等)为星型支化剂的核,通过酰胺化反应可控制备具有高结晶性、高柔韧性的多官能团长链星型支化剂,或者通过官能度≥3的环己烷烷基酸(例如环己烷四甲酸等)和长碳链内酰胺反应后作为支化剂核获得长链星型支化剂。并将两种长链星型支化剂分别以一定比例加入长碳链尼龙盐中通过熔融聚合合成长碳链星型聚酰胺,使得制备得到的长碳链聚酰胺具有流动性高、结晶速度快、抗冲击性能好等特点,且制备方法具有原料易得,过程温和可控,适合大规模工业化生产等特点,克服了现有技术中的缺陷,并在此基础上,完成了本发明。
本发明第一方面提供一种长链星型支化剂的制备方法,采用两步法可控制制备长链星型支化剂的臂长及分子量。所述长链星型支化剂的制备方法包括如下步骤:
1)将环己烷二甲酰氯与长碳链二元胺、第一缚酸剂通过界面聚合反应,制备获得低分子量聚合物;
2)将环己烷烷基酰氯与步骤1)所述的低分子量聚合物继续反应制备获得长链星型支化剂;其中,所述环己烷烷基酰氯的官能度≥3。
所述环己烷烷基酰氯为环己烷上连接有烷基酰氯基团,烷基酰氯基团的个数≥3。在一实施例中,环己烷烷基酰氯为中心对称结构;优选的,所述环己烷烷基酰氯中酰氯取代基团在环己烷的1,2,4,5位或者1,3,5位。进一步的,烷基酰氯基团例如可以是甲酰氯或乙酰氯等。更具体的,所述环己烷烷基酰氯选自1,2,4,5-环己烷四甲酰氯、1,3,5-环己烷三甲酰氯、1,3,5-环己烷三乙酰氯中的一种或多种的组合。
需要说明的是,步骤1)中界面聚合反应是长碳链二元胺在水相,环己烷二甲酰氯在有机相,可以在界面处快速反应聚合。其中,有机相例如甲苯等。
本发明所提供的长链星型支化剂的制备方法中,所述步骤1)中,所述环己烷二甲酰氯的制备方法包括将环己烷二甲酸进行酰氯化反应制备获得环己烷二甲酰氯。更具体的,所述环己烷二甲酰氯的制备方法包括向环己烷二甲酸中加入第一溶剂和第一催化剂反应后,提纯、蒸馏、干燥制备获得。
在一些实施例中,所述步骤1)中,所述环己烷二甲酸选自1,4-环己烷二甲酸;所述环己烷二甲酰氯选自1,4-环己烷二甲酰氯。
例如,向1,4-环己烷二甲酸缓慢加入溶解三光气的甲苯溶液,加入第一催化剂,在冰水混合浴中搅拌直到1,4-环己烷二甲酸反应完全,提纯后减压蒸馏、干燥得到1,4-环己烷二甲酰氯。更具体的,1,4-环己烷二甲酸缓慢加入溶解三光气的甲苯溶液,加入第一催化剂,在冰水混合浴中反应0.1~2h,0.1~1h,或1~2h。然后油浴加热中继续30~90℃,30~50℃,50~70℃,或70~90℃下继续反应0.1~2h,0.1~1h,或1~2h。
本发明所提供的长链星型支化剂的制备方法中,第一溶剂选自三光气(碳酸三氯甲基酯)的甲苯溶液;所述第一催化剂选自n,n-二甲基甲酰胺、吡啶、三乙胺、咪唑中的一种或多种的组合。
本发明所提供的长链星型支化剂的制备方法中,所述步骤1)中,所述长碳链二元胺选自壬二胺、癸二胺、十一碳二元胺、十二碳二元胺等中的一种或多种的组合。
本发明所提供的长链星型支化剂的制备方法中,所述步骤1)中,所述第一缚酸剂选自四丁基氢氧化胺,三乙胺,n,n-二异丙基乙胺,4-二甲氨基吡啶,碳酸钠,碳酸氢钠,尿素等中的一种或多种的组合。
本发明所提供长链星型支化剂的制备方法中,所述步骤1)中,所述长碳链二元胺和环己烷二甲酸的摩尔比通常情况下没有限定。例如,所述长碳链二元胺和环己烷二甲酸的摩尔比0.5~2,0.5~1,或1~2。
本发明所提供的长链星型支化剂的制备方法中,所述步骤2)是将环己烷烷基酰氯与步骤1)所述的低分子量聚合物聚合反应制备获得长链星型支化剂。其中,所述步骤2)中,所述环己烷烷基酰氯的制备方法包括将环己烷烷基酸(环己烷烷基酸的官能度≥3)进行酰氯化反应制备获得环己烷烷基酰氯。在一实施例中,所述环己烷烷基酰氯的制备方法包括向环己烷烷基酸中加入第二溶剂和第二催化剂反应后,提纯、蒸馏、干燥制备获得。
所述步骤2)中,所述环己烷烷基酸为环己烷上连接有烷基酸基团,烷基酸基团的个数为≥3。在一实施例中,环己烷烷基酸为中心对称结构;优选的,所述环己烷烷基酸中酸取代基团在环己烷的1,2,4,5位或者1,3,5位。进一步的,烷基酸基团例如可以是甲酸或乙酸等。更具体的,所述环己烷烷基酸选自1,2,4,5-环己烷四甲酸、1,3,5-环己烷三甲酸、1,3,5-环己烷三乙酸中的一种或多种的组合。
以所述环己烷烷基酸选自1,2,4,5-环己烷四甲酸为例,向1,2,4,5-环己烷四甲酸缓慢加入溶解三光气的甲苯溶液,加入第二催化剂,在冰水混合浴中搅拌直到1,2,4,5-环己烷四甲酸反应完全,提纯后减压蒸馏、干燥得到1,2,4,5-环己烷四甲酰氯。更具体的,1,2,4,5-环己烷四甲酸缓慢加入溶解三光气的甲苯溶液,加入第二催化剂,在冰水混合浴中反应0.1~2h,0.1~1h,或1~2h。然后油浴加热中继续30~90℃,30~50℃,50~70℃,或70~90℃下继续反应0.1~2h,0.1~1h,或1~2h。
本发明所提供长链星型支化剂的制备方法中,所述步骤2)中,所述第二溶剂选自三光气的甲苯溶液;所述第二催化剂选自n,n-二甲基甲酰胺、吡啶、三乙胺、咪唑中的一种或多种的组合。
本发明所提供长链星型支化剂的制备方法中,所述长碳链二元胺和环己烷烷基酸的摩尔比为0.5~4:1。在一些实施例中,长碳链二元胺和环己烷烷基酸的摩尔比例如可以为0.5~1:1,1~2:1,2~3:1,或3~4:1。具体的,长碳链二元胺和1,2,4,5-环己烷四甲酸的摩尔比为0.5~4:1,0.5~1:1,1~2:1,2~3:1,或3~4:1。
本发明所提供长链星型支化剂的制备方法中,所述第一缚酸剂和环己烷烷基酸的摩尔比为0.8~10:1。在一些实施例中,所述第一缚酸剂和环己烷烷基酸的摩尔比可以为0.8~1:1,1~2:1,2~3:1,3~4:1,4~5:1,5~6:1,6~7:1,7~8:1,8~9:1,或9~10:1。具体的,所述第一缚酸剂和1,2,4,5-环己烷四甲酸的摩尔比0.8~10:1,0.8~1:1,1~2:1,2~3:1,3~4:1,4~5:1,5~6:1,6~7:1,7~8:1,8~9:1,或9~10:1。
本发明所提供长链星型支化剂的制备方法中,所述第一催化剂和环己烷烷基酸的质量比为1:1~5。在一些实施例中,所述第一催化剂和环己烷烷基酸的质量比例如可以为1:1~2,1:2~3,1:3~4,或1:4~5。具体的,所述第一催化剂和1,2,4,5-环己烷四甲酸的质量比为1:1~5,1:1~2,1:2~3,1:3~4,或1:4~5。
本发明所提供长链星型支化剂的制备方法中,所述第二催化剂和环己烷烷基酸的质量比为1:1~5。在一些实施例中,所述第二催化剂和环己烷烷基酸的质量比例如可以为1:1~2,1:2~3,1:3~4,或1:4~5。具体的,所述第二催化剂和1,2,4,5-环己烷四甲酸的质量比为1:1~5,1:1~2,1:2~3,1:3~4,或1:4~5。
本发明所提供长链星型支化剂的制备方法中,所述低分子量聚合物的分子量为200~2000。在一些实施例中,所述低分子量聚合物的分子量可以为200~500,500~1000,1000~1500,或1500~2000。
本发明所提供长链星型支化剂的制备方法中,所述长链星型支化剂的分子量为1400~10000。在一些实施例中,所述长链星型支化剂的分子量可以为1400~2000,2000~3000,3000~4000,4000~5000,5000~6000,6000~7000,7000~8000,8000~9000,或9000~10000。
本发明所提供长链星型支化剂的制备方法中,将所述环己烷二甲酰氯与长碳链二元胺、第一缚酸剂在两相溶剂中发生界面聚合反应,反应温度为30~90℃,30~50℃,50~70℃,或70~90℃下继续搅拌0.1~2h,0.1~1h,或1~2h。然后再加入环己烷烷基酰氯,反应0.1~2h,0.1~1h,或1~2h。制备获得长链星型支化剂。
本发明第二方面提供另一种长链星型支化剂的制备方法,所述长链星型支化剂的制备方法包括:将如本发明第一方面中所述的环己烷烷基酰氯、长碳链内酰胺与第二缚酸剂反应制备获得长链星型支化剂;其中,所述环己烷烷基酰氯的官能度≥3。
所述环己烷烷基酰氯为环己烷上连接有烷基酰氯基团,烷基酰氯基团的个数为≥3。在一实施例中,环己烷烷基酰氯为中心对称结构;优选的,所述环己烷烷基酰氯中酰氯取代基团在环己烷的1,2,4,5位或者1,3,5位。进一步的,烷基酰氯基团例如可以是甲酰氯或乙酰氯等。更具体的,所述环己烷烷基酰氯选自1,2,4,5-环己烷四甲酰氯、1,3,5-环己烷三甲酰氯、1,3,5-环己烷三乙酰氯中的一种或多种的组合。
本发明所提供长链星型支化剂的制备方法中,所述环己烷烷基酰氯的制备方法包括将环己烷烷基酸(环己烷烷基酸的官能度≥3)进行酰氯化反应制备获得环己烷烷基酰氯。其中,所述环己烷烷基酸为环己烷上连接有烷基酸基团,烷基酸基团的个数为≥3。在一实施例中,环己烷烷基酸为中心对称结构;优选的,所述环己烷烷基酸中酸取代基团在环己烷的1,2,4,5位或者1,3,5位。进一步的,烷基酸基团例如可以是甲酸或乙酸等。更具体的,所述环己烷烷基酸选自1,2,4,5-环己烷四甲酸、1,3,5-环己烷三甲酸、1,3,5-环己烷三乙酸中的一种或多种的组合。
需要说明的是,本发明第一方面中关于环己烷烷基酰氯的合成方法,包括原料的选择以及原料之间的配比等限定均适用于本发明第二方面使用的环己烷烷基酰氯中。
本发明所提供长链星型支化剂的制备方法中,所述长碳链内酰胺和环己烷烷基酸的摩尔比为0.5~4:1。在一些实施例中,所述长碳链内酰胺和环己烷烷基酸的摩尔比例如可以为0.5~1:1,1~2:1,2~3:1,或3~4:1。具体的,所述长碳链内酰胺和1,2,4,5-环己烷四甲酸的摩尔比为0.5~4:1,0.5~1:1,1~2:1,2~3:1,或3~4:1。
本发明所提供长链星型支化剂的制备方法中,所述第二缚酸剂和环己烷烷基酸的摩尔比为0.8~10:1。在一些实施例中,所述第二缚酸剂和环己烷烷基酸的摩尔比可以为0.8~1:1,1~2:1,2~3:1,3~4:1,4~5:1,5~6:1,6~7:1,7~8:1,8~9:1,或9~10:1。具体的,所述第二缚酸剂和1,2,4,5-环己烷四甲酸的摩尔比0.8~10:1,0.8~1:1,1~2:1,2~3:1,3~4:1,4~5:1,5~6:1,6~7:1,7~8:1,8~9:1,或9~10:1。
本发明所提供长链星型支化剂的制备方法中,所述的长碳链内酰胺选自十一碳内酰胺和/或十二碳内酰胺。
本发明所提供长链星型支化剂的制备方法中,所述第二缚酸剂选自选自1,2,4,5-环己烷四甲酰氯、1,3,5-环己烷三甲酰氯、1,3,5-环己烷三乙酰氯中的一种或多种的组合。
本发明所提供长链星型支化剂的制备方法中,将所述环己烷烷基酰氯与长碳链内酰胺、第二缚酸剂反应,反应温度为30~90℃,30~50℃,50~70℃,或70~90℃下继续搅拌0.1~2h,0.1~1h,或1~2h。
本发明第三方面提供一种长链星型支化剂,由如本发明第一方面或本发明第二方面所述的长链星型支化剂的制备方法制备获得。
本发明第四方面提供一种长碳链星型聚酰胺,所述长碳链星型聚酰胺的原料包括长碳链尼龙盐、本发明第三方面所述的长链星型支化剂、助剂和水。
本发明所提供的长碳链星型聚酰胺中,长碳链星型聚酰胺按其重量份计,包括90~100份,90~95份,或95~100份的长碳链尼龙盐。其中,长碳链尼龙盐选自癸二酰己二胺盐、癸二酰癸二胺盐、十二烷二酰己二胺盐、十二烷二酰癸二胺盐、十二烷二酰十二烷二胺盐等中的一种或多种的组合。
本发明所提供的长碳链星型聚酰胺中,长碳链星型聚酰胺按其重量份计,包括0.1~4份,0.1~1份,1~2份,2~3份,或3~4份的长链星型支化剂。
本发明所提供的长碳链星型聚酰胺中,长碳链星型聚酰胺按其重量份计,包括40~60份,40~45份,45~50份,50~55份,或55~60份的水。
本发明所提供的长碳链星型聚酰胺中,所述助剂包括催化剂、消泡剂、抗氧剂、分子量调节剂等。
本发明所提供的长碳链星型聚酰胺中,长碳链星型聚酰胺按其重量份计,包括0.002~0.01份,0.002~0.005份,0.005~0.008份,或0.008~0.01份的第三催化剂。其中,所述第三催化剂选自次磷酸水溶液、次亚磷酸钠、次亚磷酸钾、次亚磷酸钙中的一种或多种的组合。其中次磷酸水溶液为次磷酸(50%)水溶液。
本发明所提供的长碳链星型聚酰胺中,长碳链星型聚酰胺按其重量份计,包括0.1~1份,0.1~0.3份,0.3~0.5份,0.5~0.8份,或0.8~1份的消泡剂。所述消泡剂选自聚乙二醇、聚乙二醇-丙二醇共聚物、聚二甲基硅氧烷中的一种或多种的组合。其中聚乙二醇的分子量为1800~8000,1800~3000,3000~5000,或5000~8000。
本发明所提供的长碳链星型聚酰胺中,长碳链星型聚酰胺按其重量份计,包括0.1~0.5份,0.1~0.3份,或0.3~0.5份的抗氧剂。其中,所述抗氧剂选自抗氧剂1098、抗氧剂168和抗氧剂1010等中的一种或多种的组合。
本发明所提供的长碳链星型聚酰胺中,长碳链星型聚酰胺按其重量份计,包括0.1~2份,0.1~0.5份,0.5~1份,1~1.5份,或1.5~2份的分子量调节剂。其中,所述分子量调节剂选自苯甲酸、醋酸、长链二元酸和长链二元胺等中的一种或多种的组合。
本发明第五方面提供本发明所述的长碳链星型聚酰胺的制备方法,包括将长链星型支化剂与长碳链尼龙盐以及助剂混合后进行聚合反应制备获得。
具体的,将长链星型支化剂与长碳链尼龙盐、第三催化剂、消泡剂、抗氧剂、分子量调节剂、水按本发明第三方面所述的长碳链星型聚酰胺中重量份混合,升高温度进行保压预聚合,后泄压至常压继续反应脱水。缩聚反应完成后,将物料压出、拉条、切粒、干燥得到高流动性的长链星型聚酰胺。
需要说明的是,长链星型支化剂与长碳链尼龙盐、第三催化剂、消泡剂、抗氧剂、分子量调节剂、长链星型支化剂的具体选择同本发明第四方面所述的长碳链星型聚酰胺。
综上,与现有技术相比,本发明具有以下优点:
1)本发明的优点在于本发明生产的长碳链星型聚酰胺,具有结晶速度快、流动性佳、抗冲性能好等优点。
2)本发明的高流动性高结晶性长碳链聚酰胺的制备方法,具有原料易得,聚合过程可控,适合大规模工业化生产等特点。
以下由特定的具体实施例说明本发明的实施方式,熟悉此技术的人士可由本说明书所揭露的内容轻易地了解本发明的其他优点及功效。
在进一步描述本发明具体实施方式之前,应理解,本发明的保护范围不局限于下述特定的具体实施方案;还应当理解,本发明实施例中使用的术语是为了描述特定的具体实施方案,而不是为了限制本发明的保护范围。下列实施例中未注明具体条件的试验方法,通常按照常规条件,或者按照各制造商所建议的条件。
当实施例给出数值范围时,应理解,除非本发明另有说明,每个数值范围的两个端点以及两个端点之间任何一个数值均可选用。除非另外定义,本发明中使用的所有技术和科学术语与本技术领域技术人员通常理解的意义相同。除实施例中使用的具体方法、设备、材料外,根据本技术领域的技术人员对现有技术的掌握及本发明的记载,还可以使用与本发明实施例中所述的方法、设备、材料相似或等同的现有技术的任何方法、设备和材料来实现本发明。
实施例1
向三口烧瓶中加入500ml甲苯,30g三光气,持续搅拌1h,转移三口烧瓶于冰水混合浴中,缓慢加入26g1,2,4,5-环己烷四甲酸和1mln,n-二甲基甲酰胺继续反应2h,转移至油浴中,50℃下继续反应4h,,减压蒸馏得到1,2,4,5-环己烷四甲酰氯。在同样的条件下用1,4-环己烷二甲酸制备1,4-环己烷二甲酰氯。加入174.2g1,4-环己烷二甲酰氯、175.5g癸二胺、550ml去离子水,500ml甲苯,168g四丁基氢氧化铵,在40℃下继续搅拌30min,加入31.0g1,2,4,5-环己烷四甲酰氯,搅拌10min。降温,过滤,真空干燥得到长链星型支化剂s1。
向高压反应釜中依次投入10kg的癸二酰癸二胺盐,其中癸二酰癸二胺盐是由等摩尔的癸二酸和癸二胺通过中和反应得到。再依次加376g长链星型支化剂s1,48g癸二胺,97g苯甲酸,1g催化剂次亚磷酸钠,30g消泡剂carbowaxpeg8000,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。熔体粘度如图1。
实施例2
向三口烧瓶中加入500ml甲苯,15g三光气,持续搅拌1h,转移三口烧瓶于冰水混合浴中,缓慢加入13g1,2,4,5-环己烷四甲酸和0.5mln,n-二甲基甲酰胺继续反应2h,转移至油浴中,50℃下继续反应4h,,减压蒸馏得到1,2,4,5-环己烷四甲酰氯。同样方法用1,4-环己烷二甲酸得到二官能度的1,4-环己烷二甲酰氯。加入310g1,4-环己烷二甲酰氯、287g癸二胺、550ml去离子水,500ml甲苯,116g尿素,在70℃下继续搅拌1h,再加入31g1,2,4,5-环己烷四甲酰氯,搅拌10min得到长链星型支化剂s2。
向高压反应釜中依次投入10kg的癸二酰癸二胺盐,490g长链星型支化剂s2,48g癸二胺,97g苯甲酸,1g催化剂次亚磷酸钠,30g消泡剂carbowaxpeg8000,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。
对比例1
向高压反应釜中依次投入10kg的癸二酰癸二胺盐,21g1,2,4,5-环己烷四甲酸二酐(星型支化剂s3),48g癸二胺,97g苯甲酸,1g催化剂,30g消泡剂,6.7kg水。反复使用氮气置换釜内气氛,提高物料温度至80℃,反应30min。继续提升釜内温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。熔体粘度如图1。
实施例3
向三口烧瓶中加入500ml甲苯,30g三光气,持续搅拌1h,转移三口烧瓶于冰水混合浴中,缓慢加入26g1,2,4,5-环己烷四甲酸和10mln,n-二甲基甲酰胺继续反应2h,转移至油浴中,50℃下继续反应4h,,减压蒸馏得到1,2,4,5-环己烷四甲酰氯。使用相同方法将1,4-环己烷二甲酸酰氯化。加入261g1,2,4,5-环己烷二甲酰氯、287g十二烷二胺、550ml去离子水,500ml甲苯,168g四丁基氢氧化铵,在40℃下继续搅拌1h,再加入31g1,4-环己烷四甲酰氯,搅拌10min。降温,过滤,真空干燥得到长链星型支化剂s4。
向高压反应釜中依次投入10kg的十二烷二酰十二烷二胺盐,456g长链星型支化剂s4,56g十二烷二胺,97g苯甲酸,1g催化剂次亚磷酸钠,30g消泡剂carbowaxpeg8000,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。
实施例4
向三口烧瓶中加入500ml甲苯,30g三光气,持续搅拌1h,转移三口烧瓶于冰水混合浴中,缓慢加入26g1,2,4,5-环己烷四甲酸和1mln,n-二甲基甲酰胺继续反应2h,转移至油浴中,50℃下继续反应4h,,减压蒸馏得到1,2,4,5-环己烷四甲酰氯。加入73g十二内酰胺、400ml去离子水,100ml甲苯,168g四丁基氢氧化铵,在40℃下继续搅拌1h,降温,过滤,真空干燥得到长链星型支化剂s5。
向高压反应釜中依次投入10kg的十二烷二酰癸二胺盐,101g长链星型支化剂s5,48g癸二胺,97g苯甲酸,1g催化剂次亚磷酸钠,30g消泡剂carbowaxpeg8000,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。
实施例5
向三口烧瓶中加入500ml甲苯,30g三光气,持续搅拌1h,转移三口烧瓶于冰水混合浴中,缓慢加入26g1,2,4,5-环己烷四甲酸和1mln,n-二甲基甲酰胺继续反应2h,转移至油浴中,50℃下继续反应4h,减压蒸馏得到1,2,4,5-环己烷四甲酰氯。在同样的条件下用1,4-环己烷二甲酸制备1,4-环己烷二甲酰氯。加入174.2g1,4-环己烷二甲酰氯、175.5g癸二胺、550ml去离子水,500ml甲苯,168g四丁基氢氧化铵,在40℃下继续搅拌30min,加入31.0g1,2,4,5-环己烷四甲酰氯,搅拌10min。降温,过滤,真空干燥得到长链星型支化剂s6。
向高压反应釜中依次投入10kg的癸二酰癸二胺盐,188g长链星型支化剂s6,24g癸二胺,243g苯甲酸,1g催化剂次亚磷酸钠,30g消泡剂carbowaxpeg8000,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。
实施例6
向三口烧瓶中加入500ml甲苯,30g三光气,持续搅拌1h,转移三口烧瓶于冰水混合浴中,缓慢加入26g1,2,4,5-环己烷四甲酸和1mln,n-二甲基甲酰胺继续反应2h,转移至油浴中,50℃下继续反应4h,减压蒸馏得到1,2,4,5-环己烷四甲酰氯。在同样的条件下用1,4-环己烷二甲酸制备1,4-环己烷二甲酰氯。加入174.2g1,4-环己烷二甲酰氯、175.5g癸二胺、550ml去离子水,500ml甲苯,168g四丁基氢氧化铵,在40℃下继续搅拌30min,加入31.0g1,2,4,5-环己烷四甲酰氯,搅拌10min。降温,过滤,真空干燥得到长链星型支化剂s7。
向高压反应釜中依次投入10kg的癸二酰癸二胺盐,564g长链星型支化剂s7,48g癸二胺,24g苯甲酸,1g催化剂次亚磷酸钠,30g消泡剂carbowaxpeg8000,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。
对比例2
向高压反应釜中依次投入10kg的癸二酰癸二胺盐,41g聚醚胺t403,37g癸二酸,1g催化剂次亚磷酸钠,30g消泡剂carbowaxpeg8000,97g苯甲酸,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。熔体粘度如图1。
对比例3
向高压反应釜中依次投入10kg的癸二酰癸二胺盐,19g均苯三甲酸,32g癸二胺,1g催化剂次亚磷酸钠,30g消泡剂carbowaxpeg8000,97g苯甲酸,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。
实施例7
表1和表2为上述实施例中制备的星型长碳链尼龙的力学和热力学数据。c1为不加入星型支化剂制备的线性聚酰胺。向高压反应釜中依次投入10kg的癸二酰癸二胺盐,97g苯甲酸,1g催化剂,30g消泡剂,6.7kg水。反复使用氮气置换釜内气氛后提升釜内物料温度,直至釜内压力达到1.8mpa。打开排水阀,不断排水提高物料温度至245℃。保压1.5h后降低釜内压力至大气压,在微正压氮气或微负压真空下继续反应增粘1h,出料,切粒,干燥,打包。
表1
其中,材料抗拉强度、拉伸模量和缺口冲击强度测试均在23℃、rh=50%的环境下进行,拉伸速率为10mm/min。黏数为使用间甲酚溶解长碳链星型聚酰胺后,浓度为0.005g/ml的溶液在23℃时测得的相对粘度。等温结晶时间的测定先将材料以20℃/min升温速率升温至250℃,恒温3min消除热历史,迅速降低至测试温度进行等温结晶操作,记录完成结晶完成过程所需的半周期t1/2。聚合物的熔体流动性数据在2.16kg、230℃的操作条件测试得到。其中,抗拉强度、拉伸模量测试采用的标准是iso527。缺口冲击强度测试采用的标准为iso179。熔融指数测试采用的标准为iso1133。
表2
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



