
 商标分类
商标分类  商标转让
商标转让 
提高原油采收率的方法与流程
 2021-02-02 15:02:17|
2021-02-02 15:02:17| 344|
344| 起点商标网
起点商标网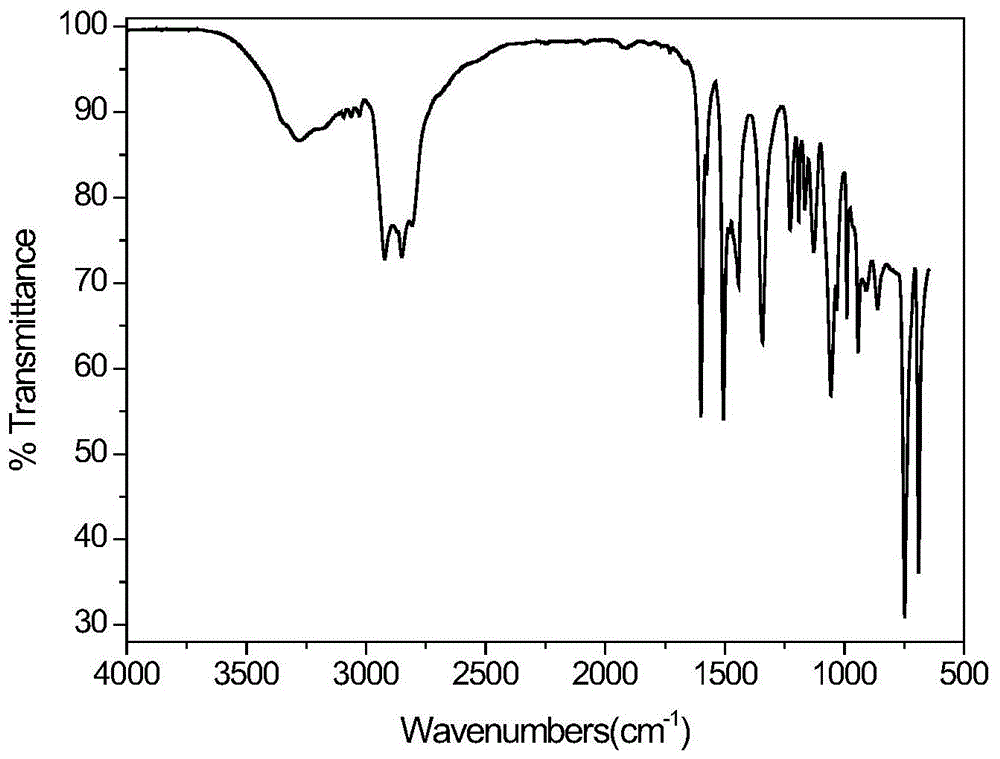
本发明涉及一种提高原油采收率的方法。
背景技术:
化学驱是通过水溶液中添加化学剂,改变注入流体的物理化学性质和流变学性质以及与储层岩石的相互作用特征而提高采收率的一种强化措施,在我国得以快速发展,其主要原因是我国储层为陆相沉积非均质性较强,陆相生油原油黏度较高,在eor方法中更适合于化学驱。
耐温抗盐聚合物的使用是提高原油采收率的一个关键因素。早期常用于强化石油开采(eor)的聚合物驱的工业产品仅有部分水解聚丙烯酰胺(hpam),它依赖于高分子量和聚合物分子链上的离子和强极性侧基的排斥作用达到增黏效果。但是,高分子量聚合物当受到较大的拉伸和剪切应力时,易于发生机械降解而丧失黏度,在低渗透率岩层中注入聚合物时尤为明显。水溶液中的阳离子,尤其是二价离子,会屏蔽聚合物中的离子基团,使聚合物分子链卷曲,流体力学体积减小甚至沉淀,从而使增黏性大大降低。当地层油层温度较高时(>93℃),聚丙烯酰胺(pam)中的酰胺基在高温水溶液中易水解,使聚合物溶液的抗盐性急剧下降。近年来,有关耐温抗盐型聚丙烯酰胺的研究主要通过在聚合物主链上引入大侧基或刚性侧基提高聚合物的热稳定性、引入抑制水解的单体或对盐不敏感的单体进行共聚来提高聚合物的耐水解及抗盐性能,或通过疏水基团的疏水缔合作用来改善聚合物的耐温抗盐性能。
表面活性剂作为化学驱的一个重要组成,根据其化学组成及分子结构不同,可以分为离子型与非离子型两大类。目前三次采油研究中所用表面活性剂的种类以阴离子型最多,其次是非离子型和两性离子型,应用最少的是阳离子型。美孚石油公司的专利us3927716、us4018281、us4216097相继报道了采用碱水驱油、表面活性剂或碱水驱油及使用两性离子表面活性剂驱油的结果,采用的两性离子表面活性剂为不同链长的羧酸或磺酸盐型甜菜碱表面活性剂,在总矿化62000~160000mg/l,钙镁离子1500~18000mg/l的模拟盐水中,对德克萨斯南部原油的界面张力达10-1~10-4mn/m。使用阳离子表面活性剂的亦有报道,如中国专利cn1528853、cn1817431、cn1066137等相继报道了双酰胺型阳离子型、含氟阳离子型及含吡啶基阳离子双子表面活性剂,但由于阳离子具有吸附损耗大、成本高等缺点,限制了其在油田现场的使用。
不同类型表面活性剂相互复配后往往可以克服单一表面活性剂的缺点,发挥各组分的优点,从而赋予表面活性剂组合物更加优越的性能。中国专利cn1458219a公开了一种三次采油应用的表面活性剂/聚合物二元超低界面张力复合驱配方,其中使用的表面活性剂是石油磺酸盐或以石油磺酸盐为主剂加稀释剂和其它表面活性剂复配的表面活性剂组合物,其组份的重量百分比为石油磺酸盐50~100%,烷基磺酸盐0~50%,羧酸盐0~50%,烷基芳基磺酸盐0~35%,低碳醇0~20%,该表面活性剂体系过于复杂。美国德克萨斯大学的专利us8211837报道了采用简单廉价的线性醇在高温下催化二聚反应得到支链化的长碳醇,与环氧丙烷、环氧乙烷聚合后进行硫酸酯化反应,相对于昂贵的磺酸盐型表面活性剂,低成本合成了大亲水基聚醚硫酸盐表面活性剂,由于大亲水基团的存在,从而使得该硫酸盐表面活性剂在碱性条件下高温稳定性能优良,0.3%的支链醇聚醚硫酸盐(c32-7po-6eo硫酸盐)与0.3%的内烯烃磺酸盐(c20~24ios)盐水溶液在85℃与相同量的原油混合,其增溶参数为14。美孚石油公司的专利us4370243报道了采用油溶性的醇、磺酸甜菜碱及季铵盐组成的驱油体系,该体系既可起到表面活性剂的作用,也可起到流度控制剂的作用,其中季铵盐为亲油基碳链长为16~20的阳离子表面活性剂,采用2%的十八烷基二羟乙基丙基磺酸盐甜菜碱与1.0%的正己醇作为驱油剂,注入1.9pv后,原油即可100%驱出,但表面活性剂的吸附损耗较大达到6mg/g,在此基础上加入价格相对低廉的2.0%四乙基溴化铵作为牺牲剂以降低表面活性剂的吸附量。
不同类型的表面活性剂之间会存在协同作用,特别是电性相反的表面活性剂复配具有极高的表界面活性,因而具有非常广阔的应用前景。如阎云等(见《物理化学学报》2002年第9期,830~834)研究了规则溶液理论应用于异电性传统表面活性剂十二烷基硫酸钠(sds)与bola型两亲分子[(me)3n+(ch2)6oc6h4o(ch2)6n+(me)3]2br-的相互作用,bola分子与sds混合体系中的协同作用主要是由亲水基之间的静电相互作用产生的,而bola分子结构中疏水部分对相互作用没有显著影响,中国石化胜利油田分公司曹绪龙(见《物理化学学报》2014年第7期,1297~1302)研究了阴阳离子表面活性剂混合体系对原油的乳化及增黏行为,对油水体积比、浓度、温度、ph值、离子强度对乳化增黏的影响进行了系统的研究,得到了具有最佳增黏效果的配方体系,与原油黏度相比,实现了80倍左右的黏度上升。
表面活性剂制备方法中,阴-非离子表面活性剂往往是由非离子表面活性剂通过羧甲基化改性而来的,如美国专利us4818440报道了脂肪酰胺聚氧乙烯醚乙酸的制备及应用,其中羧甲基化反应采用的是分批次加入氢氧化钠和氯乙酸钠固体,最终聚醚转化率达到75~80%,合成的粗产品经过酸化提纯得到纯度较高的脂肪酰胺聚氧乙烯醚乙酸,作为主要成分应用于香波等日化产品中。该报道中,聚醚羧酸盐需经过提纯,合成工艺复杂,产生的三废多,制备成本较高,且未涉及在提高原油采收率方面的应用。
上述国内外研究结果表明,降低驱油剂的制备成本、提高驱油剂的驱油效率是提高原油采收率的关键。本发明所述的正是这种在油藏条件下结构稳定的表面活性剂组合物及驱油剂和制备和及提高原油采收率的方法。
技术实现要素:
本发明所要解决的技术问题之一是现有技术中的含有表面活性剂的驱油剂体系存在的表面活性剂对原油乳化增容能力差、驱油效率低,以及耐温抗盐性能差、吸附滞留量大而无法满足高温高盐油藏驱油需要的问题的问题,提供一种提高原油采收率的方法,该方法采用的驱油剂中含有新的驱油用表面活性剂组合物,该表面活性剂组合物的水溶液能很好乳化原油,具有较强的乳化增溶原油能力,最大增溶参数为17.5~24.8,且油水界面张力可达10-3~10-4mn/m,具有耐温抗盐性能好、吸附滞留量低的优点,从而有效提高了原油驱油效率,具有很好的提高采收率应用前景。
本发明所要解决的技术问题之二是提供一种与解决上述技术问题之一相对应的驱油用表面活性剂组合物。
本发明所要解决的技术问题之三是提供一种与解决上述技术问题之二相对应的驱油用表面活性剂组合物的制备方法。
本发明所要解决的技术问题之四是提供一种与解决上述技术问题之二相对应的驱油用表面活性剂组合物的应用方法。
本发明所要解决的技术问题之五是提供一种与解决上述技术问题之一相对应的驱油剂,该驱油剂含有所述驱油用表面活性剂组合物。
本发明所要解决的技术问题之六是提供一种与解决上述技术问题之五相对应的驱油剂的制备方法。
为了解决上述技术问题之一,本发明采用的技术方案如下:一种提高原油采收率的方法,包括以下步骤:
(1)将驱油剂与水混合得到驱油体系;
(2)将所述驱油体系与含油地层接触,将所述含油地层中的原油驱替出来;
其中,所述驱油剂,以质量份数计,包括:
(1)1份表面活性剂组合物;
(2)0~20份聚合物且大于0份聚合物;
所述表面活性剂组合物,以摩尔份数计,包括以下组分:
(1)1份季铵盐表面活性剂;
(2)0.1~30份含胺基聚醚的表面活性剂;
其中,所述季铵盐表面活性剂的分子通式为式(i)所示:
式(i)中,r1、r2任意选自氢、(chr')coh、(chr')dch3、苯基、取代苯基或苄基中的一种,r4为氢、c2~c32的烃基或取代烃基、(chr”)coh、苯基、取代苯基或苄基中的一种,r3为氢、c2~c32的烃基或取代烃基、(chr”')eoh、卤素、氨基中的一种,r'、r”、r”'独立选自h、ch3或c2h5中的一种,c为1~4中的任一整数、d为0~5中的任一整数、e为0~4中的任一整数;xj-为负电荷数为j的阴离子或阴离子基团;
所述含胺基聚醚的表面活性剂的分子通式为式(ii)所示:
式(ii)中,r5为c8~c30的直链或支链的饱和及不饱和烷基或由c4~c20直链或支链的饱和及不饱和烃基或枯基(c6h5c(ch3)2)取代的苯环或萘环,或r5n为松香胺根;r1、r2、r3或r4独立选自0~50,但r1和r2、r3和r4不能同时为0;s1和s2独立选自0~100,但s1和s2不能同时为0;y为h或r6z;y'为h或r'6z′;r6和r′6独立选自c1~c5的亚烷基或羟基取代亚烷基中的至少一种;z和z′独立选自cooa、so3b或氢中的一种,a、b任意选自阳离子或阳离子基团。
上述技术方案中,所述聚合物优选为阴离子型聚丙烯酰胺、耐温抗盐改性聚丙烯酰胺、疏水缔合聚丙烯酰胺或聚合物微球中的至少一种;进一步优选所述耐温抗盐改性聚丙烯酰胺分子链中优选包括丙烯酰胺结构单元、2-丙烯酰胺基-2-甲基丙磺酸结构单元,丙烯酰胺结构单元、2-丙烯酰胺基-2-甲基丙磺酸结构单元摩尔比优选为(0.1~40)∶1,黏均分子量优选为800~2500万;所述疏水缔合聚合物分子链中优选包括丙烯酰胺结构单元、耐温抗盐单体结构单元与疏水单体结构单元,丙烯酰胺结构单元、耐温抗盐单体结构单元与疏水单体结构单元的摩尔比优选为1:(0.1~40):(0.001~0.05),黏均分子量优选为500~2500万;所述驱油剂中,进一步优选还包括0~30份碱,更优选所述表面活性剂组合物与聚合物与碱的质量比优选为1∶(0.1~2):(0~5)。
上述技术方案中,所述a、b任意选自氢离子、碱金属阳离子或者由式nr7(r8)(r9)(r10)所示基团中的至少一种,其中,r7、r8、r9、r10为独立选自h、(chr0)foh或(chr0)gch3中的一种,r0为h、ch3或c2h5中的一种,f为1~4中的任意整数,g为0~5中的任意整数。
上述技术方案中,所述r1、r2任意选自氢、甲基、乙基、羟乙基、羟丙基、苯基、苄基中的一种;r4为氢、c8~c24的烃基或取代烃基、甲基、乙基、羟乙基、羟丙基、苯基、苄基中的一种,r3为氢、c8~c24的烃基或取代烃基、氢、甲基、乙基、苯基、羟基、氨基中的一种;r'、r”、r”'、r0独立选自h或ch3;c=1~2,d=0~1,e=0~1,f=1~2,g=0~1;所述r5为c12~c24的烃基或取代烃基或由c4~c20直链或支链的饱和及不饱和烃基或枯基(c6h5c(ch3)2)取代的苯环或萘环,或r5n为松香胺根;r6和r′6为c1~c3的亚烷基或氢中的一种;所述r1+r2=1~10,r3+r4=1~10,s1+s2=1~20。
上述技术方案中,所述j=1~3中的任一整数;j=1时,x-为cl-、br-、i-、ch3oso3-、hco3-、hcoo-、ch3coo-、c2h5coo-、c3h7coo-、hoc6h4coo-、c6h5so3-或ch3c6h4so3-中的一种,进一步优选为cl-、br-、hco3-、ch3coo-、hoc6h4coo-、c6h5so3-或ch3c6h4so3-中的一种;j=2时,x2-为so42-、hpo42-、(coo-)2、ch2(coo-)2、c2h4(coo-)2、c2h2(coo-)2或c6h4(coo-)2中的一种;进一步优选为so42-、(coo-)2、c2h2(coo-)2或c6h4(coo-)2中的一种;j=3时,x3-为po43-或ch2(coo-)ch(oh)(coo-)ch2(coo-)中的一种,进一步优选为ch2(coo-)ch(oh)(coo-)ch2(coo-)。
上述技术方案中,所述表面活性剂组合物还包括:
(3)0~20份小分子醇;
(4)0~20份小分子胺;
(5)0~10份盐;
(6)0~10份无机碱;
其中,所述小分子醇选自c1~c8的脂肪醇;所述小分子胺选自c1~c8的伯胺、仲胺或叔胺中的至少一种;盐选自金属卤化物、羟基取代的羧酸盐中至少一种;无机碱选自碱金属氢氧化物、碱金属碳酸盐或碱金属碳酸氢盐中的至少一种;进一步优选:所述季铵盐表面活性剂、含胺基聚醚的表面活性剂、小分子醇、小分子胺、盐和碱的的摩尔比优选为1:(0.2~20):(0~15):(0~15):(0~5):(0~5)。
为解决上述技术问题之二,本发明所采用的技术方案如下:一种表面活性剂组合物,以摩尔份数计,包括以下组分:
(1)1份季铵盐表面活性剂;
(2)0.1~30份含胺基聚醚的表面活性剂;
其中,所述季铵盐表面活性剂的分子通式为式(i)所示:
式(i)中,r1、r2任意选自氢、(chr')coh、(chr')dch3、苯基、取代苯基或苄基中的一种,r4为氢、c2~c32的烃基或取代烃基、(chr”)coh、苯基、取代苯基或苄基中的一种,r3为氢、c2~c32的烃基或取代烃基、(chr”')eoh、卤素、氨基中的一种,r'、r”、r”'独立选自h、ch3或c2h5中的一种,c为1~4中的任一整数、d为0~5中的任一整数、e为0~4中的任一整数;xj-为负电荷数为j的阴离子或阴离子基团;
所述含胺基聚醚的表面活性剂的分子通式为式(ii)所示:
式(ii)中,r5为c8~c30的直链或支链的饱和及不饱和烷基或由c4~c20直链或支链的饱和及不饱和烃基或枯基(c6h5c(ch3)2)取代的苯环或萘环,或r5n为松香胺根;r1、r2、r3或r4独立选自0~50,但r1和r2、r3和r4不能同时为0;s1和s2独立选自0~100,但s1和s2不能同时为0;y为h或r6z;y'为h或r'6z′;r6和r′6独立选自c1~c5的亚烷基或羟基取代亚烷基中的至少一种;z和z′独立选自coom、so3n或氢中的一种,m、n任意选自阳离子或阳离子基团。
上述技术方案中,r1、r2优选为氢、甲基、乙基、羟乙基、羟丙基、苯基、苄基中的一种。
上述技术方案中,r3优选为氢、c8~c24的烃基或取代烃基、氢、甲基、乙基、苯基、羟基、氨基、羧酸基或磺酸基中的一种。
上述技术方案中,r4优选为氢、c8~c24的烃基或取代烃基、甲基、乙基、羟乙基、羟丙基、苯基、苄基中的一种。
上述技术方案中,r'、r”、r”'优选为h或ch3。
上述技术方案中,优选c=1~2、d=0~1、e=0~1。
上述技术方案中,m、n优选为氢、碱金属阳离子或者由式nr7(r8)(r9)(r10)所示基团中的至少一种。
上述技术方案中,r7、r8、r9、r10优选为h、(chr0)foh或(chr0)gch3中的一种。
上述技术方案中,r0优选为h、ch3或c2h5中的一种。
上述技术方案中,优选f=1~2,g=0~1。
上述技术方案中,y-优选为cl-、br-、i-、ch3oso3-、hco3-、hcoo-、ch3coo-、c2h5coo-、c3h7coo-、hoc6h4coo-、c6h5so3-或ch3c6h4so3-中的一种,y2-优选为so42-、hpo42-、(coo-)2、ch2(coo-)2、c2h4(coo-)2、c2h2(coo-)2或c6h4(coo-)2中的一种,y3-优选为po43-或ch2(coo-)ch(oh)(coo-)ch2(coo-)中的一种。
上述技术方案中,y-更优选为cl-、br-、hco3-、ch3coo-、hoc6h4coo-、c6h5so3-或ch3c6h4so3-中的一种,y2-更优选为so42-、(coo-)2、c2h2(coo-)2或c6h4(coo-)2中的一种,y3-更优选为ch2(coo-)ch(oh)(coo-)ch2(coo-)。
上述技术方案中,r5优选为c12~c24的烃基或取代烃基或由c4~c20直链或支链的饱和及不饱和烃基或枯基(c6h5c(ch3)2)取代的苯环或萘环,或r5n为松香胺根。
上述技术方案中,r6和r′6优选为c1~c3的亚烷基或氢。
上述技术方案中,优选r1+r2=1~10,r3+r4=1~10,s1+s2=1~20。
上述技术方案中,所述表面活性剂组合物,以摩尔份数计,优选还包括以下组分:
(3)小分子醇;
(4)小分子胺;
(5)盐;
(6)无机碱;
其中,所述季铵盐表面活性剂、含胺基聚醚的表面活性剂、小分子醇、小分子胺、盐和碱的的摩尔比为1:(0.1~30):(0~20):(0~20):(0~10):(0~10);所述小分子醇选自c1~c8的脂肪醇;所述小分子胺选自c1~c8的伯胺、仲胺或叔胺中的至少一种;盐选自金属卤化物、羟基取代的羧酸盐中至少一种;无机碱选自碱金属氢氧化物、碱金属碳酸盐或碱金属碳酸氢盐中的至少一种进一步优选:所述季铵盐表面活性剂、含聚醚片段的表面活性剂、小分子醇、小分子胺、盐和碱的的摩尔比优选为1:(0.2~20):(0~15):(0~15):(0~5):(0~5)。上述技术方案中,优选小分子醇为c1~c5的脂肪醇。
上述技术方案中,优选小分子胺为c1~c5的脂肪胺。
上述技术方案中,金属卤化物优选为碱金属卤化物,进一步优选为氯化钠、氯化钾、溴化钠、溴化钾中的一种;羟基取代的羧酸盐优选为羟基乙酸钠、羟基乙酸钾中的一种。
上述技术方案中,优选无机碱为碱金属氢氧化物、碳酸盐或碳酸氢盐。
上述技术方案中,式(i)表示的阳离子表面活性剂的核心在于结构中的阳离子部分,yj-没有特别限制,只要能够使与式(i)中的阳离子部分构成电中性体系的阴离子均适用本发明。简单阴离子的例子例如,yj-可以是j=1的无机阴离子(例如氯离子、溴离子或氢氧根离子、磷酸二氢根等)、j=1的有机阴离子(例如醋酸根等一元羧酸根),可以是j=2的无机阴离子(例如硫酸根、磷酸氢二根等)、j=2的有机阴离子(例如酒石酸根、邻苯二甲酸根、马来酸根);还可以是j>2的多价无机或有机阴离子,例如磷酸根、柠檬酸根。除了上述简单阴离子以外,还包括多聚阴离子(例如三聚磷酸根、多聚磷酸根等)、聚合阴离子(例如聚丙烯酸根)等。但至少从制备方法简便程度考虑,yj-优选氯离子、溴离子氢氧根或乙酸根离子。
在未发生离子化反应的情形下,式(i)表示的
本发明表面活性剂组合物关键有效成分是式(i)所示季铵盐表面活性剂和式(ii)所示含聚醚片段的表面活性剂,可以将所述和/与所述盐和醇按所需的比例混合而得,优选以下述用于解决技术问题之三的技术方案获得。
为解决上述技术问题之三,本发明所采用的技术方案如下:一种解决上述技术问题之二所述的表面活性剂组合物的制备方法,包括以下步骤:
(a)季铵盐表面活性剂的制备:
将
(b)表面活性剂组合物的制备:
①在碱性催化剂存在下,r5nh2与所需量环氧乙烷、环氧丙烷、环氧乙烷反应得到聚醚化合物;将所需量的聚醚化合物与步骤(a)得到的季铵盐表面活性剂水溶液或小分子醇水溶液混合,得到所述的表面活性剂组合物;
或通过②进一步反应得到所述表面活性剂组合物:
②将步骤①得到的聚醚化合物与离子化试剂y0r6z或y0r'6z'以及碱金属氢氧化物或碱金属醇盐以摩尔比1:(0.1~20):(0.1~20)混合,搅拌下于反应温度50~120℃反应3~15小时,按所需摩尔比加入步骤(a)得到的季铵盐表面活性剂水溶液或小分子醇水溶液,升温至40~100℃搅拌1~5小时,得到所需的表面活性剂组合物驱油剂;其中,y0选自氯、溴或碘;
或:②将步骤①得到的聚醚化合物与离子化试剂y0r6coor0或y0r'6coor0以及碱金属氢氧化物或碱金属醇盐以摩尔比1:(0.1~20):(0.1~20)混合,搅拌下于反应温度50~120℃反应3~15小时,继续加入水进行皂化反应,回流1~10小时后,按所需摩尔比加入步骤(a)得到的季铵盐表面活性剂水溶液或小分子醇水溶液,升温至40~100℃搅拌1~5小时,得到所需的表面活性剂组合物;其中,y0选自氯、溴或碘,r0选自c1~c8的烷基。
上述技术方案中,步骤(a)中压力优选为0.1~0.6mpa。
上述技术方案中,步骤(a)中小分子醇优选为c1~c4的脂肪醇。
上述技术方案中,步骤(b)①所述的反应温度优选为120~160℃,碱性催化剂优选为氢氧化钾或无水碳酸钾中的至少一种,压力优选为0.30~0.60mpa表压。
上述技术方案中,步骤(b)②所述碱金属氢氧化物优选为氢氧化钾或氢氧化钠中的至少一种,聚醚化合物与离子化试剂以及碱金属氢氧化物或碱金属醇盐的摩尔比优选为1:(2~10):(2~10),r0优选为c1~c4的烷基。
y0r6z和y0r’6z′的例子有但不限于氯乙酸、氯乙酸钠、1-氯-2-羟基丙磺酸钠等。
y0r6coor0和y0r'6coor0的例子有但不限于氯乙酸酯(例如氯乙酸乙酯)、溴乙酸酯(例如溴乙酸乙酯)等。
为了解决上述技术问题之四,本发明采取的技术方案如下:一种解决上述技术问题之二所述技术方案中任一所述的表面活性剂组合物在油田驱油中的应用。
上述技术方案中,所述驱油剂可以根据现有技术加以应用,可以单独使用,也可以与油田常用助剂复配使用;作为优选方案:所述应用优选油藏的地层盐水的总矿化度5000~200000mg/l,其中ca2++mg2+为10~15000mg/l、hco3-为0~2000mg/l;原油黏度为1.0~200.0mpa.s;地层温度为50~120℃。
本发明制备的表面活性剂组合物,由于含胺基聚醚的表面活性剂与季铵盐中的苯胺结构之间呈现协同相互作用,表现在表面活性的增加、临界胶束浓度的下降、增溶原油能力的提升等方面,产生比单一表面活性剂更高的表面活性和低的临界胶束浓度,因此,该驱油剂具有优异的乳化原油的能力和界面效率,可解决油田现场使用过程中表面活性剂对原油乳化增溶能力差而无法达到很好的洗油效率,同时超高的界面效率可以保证低浓度表面活性剂仍可保持超低的油水界面张力,从而能够提高驱油效率。另外,本发明采用的表面活性剂组合物制备方法,由于高纯度离子型表面活性剂价格较高,往往需经过萃取、柱层析等复杂的提纯步骤才能得到,从而大大增加了驱油用表面活性剂的制备成本。采用聚醚与卤代羧酸盐或卤代羧酸酯在碱金属氢氧化物或碱金属醇盐催化下生成聚醚羧酸盐或聚醚羧酸酯,无需分离或直接进行皂化反应得到聚醚羧酸盐,加入所需量的含苯胺结构化合物水或小分子醇水溶液混合,体系中的小分子醇或胺可与表面活性剂在界面形成复合膜,分配至油水两相,改善油相和水相的性质,有利于油水界面张力的而降低和微乳液的形成,生成的无机盐对界面性能有促进作用亦无需去除,可能过量的碱金属氢氧化物还可以中和原油中的酸性物质形成皂进一步提高表面活性剂对原油的乳化增溶能力,提高驱油剂的洗油效率,实现了表面活性剂的绿色生产。
本发明中涉及到表面活性剂组合物含量或者浓度的场合,均指含有上述技术方案中分子通式(i)和分子通式(ii)组份的总浓度。
为了解决上述技术问题之五,本发明采用的技术方案为:一种驱油剂,以质量份数计,包括以下组分:
(1)1份上述解决技术问题之二所述的技术方案中任一所述的表面活性剂组合物或由上述解决技术问题之三所述的技术方案中任一所述的制备方法制得的表面活性剂组合物;
(2)0~20份聚合物且大于0份聚合物;
(3)0~30份碱。
上述技术方案中,所述聚合物没有严格限制,可以是本领域技术人员所熟知的各种用于油田采油的聚合物,例如但不限定选自黄原胶、羟甲基纤维素、羟乙基纤维素、阴离子型聚丙烯酰胺、耐温抗盐改性聚丙烯酰胺、疏水缔合聚合物、聚合物微球中的至少一种。
上述技术方案中,所述耐温抗盐改性聚丙烯酰胺优选分子链中包括丙烯酰胺结构单元、耐温抗盐单体结构单元,丙烯酰胺结构单元、耐温抗盐单体结构单元摩尔比为(0.1~40)∶1,黏均分子量为800~2500万,进一步,所述耐温抗盐单体优选为2-丙烯酰胺基-2-甲基丙磺酸;所述疏水缔合聚合物分子链中包括丙烯酰胺结构单元、耐温抗盐单体结构单元与疏水单体结构单元,丙烯酰胺结构单元、耐温抗盐单体结构单元与疏水单体结构单元的摩尔比为1:(0.1~40):(0.001~0.05),黏均分子量为500~2500万。
上述技术方案中,所述疏水缔合聚合物优选由丙烯酰胺、耐温抗盐单体或疏水单体共聚而成;所述耐温抗盐改性聚丙烯酰胺优选由丙烯酰胺、耐温抗盐单体共聚而成;耐温抗盐单体或疏水单体可以是本领域技术人员所熟知的含有大侧基或刚性侧基的单体(如苯乙烯磺酸、n-烷基马来酰亚胺、丙烯酰胺基长链烷基磺酸、长链烷基烯丙基二甲基卤化铵、3-丙烯酰胺基-3-甲基丁酸等)、含耐盐基团的单体(如2-丙烯酰胺基-2-甲基丙磺酸)、含耐水解基团的单体(如n-烷基丙烯酰胺)、含可抑制酰胺基水解的基团的单体(如n-乙烯吡咯烷酮)、含疏水基团的单体等中的至少一种,耐温抗盐单体优选为2-丙烯酰胺基-2-甲基丙磺酸,疏水单体优选为2-丙烯酰胺基十二烷基磺酸。
上述技术方案中,所述的疏水缔合聚合物中丙烯酰胺与耐温抗盐单体与疏水单体的摩尔比优选为1:(0.1~40):(0.001~0.05),黏均分子量为500~2500万;更优选为丙烯酰胺与耐温抗盐单体与疏水单体的摩尔比为1∶(0.1~20)∶(0.001~0.01),黏均分子量为1200~2200万。
上述技术方案中,所述耐温抗盐改性聚丙烯酰胺中丙烯酰胺与耐温抗盐单体的摩尔优选比为(0.1~40)∶1。
上述技术方案中,所述的疏水缔合聚合物优选丙烯酰胺、2-丙烯酰胺基-2-甲基丙磺酸和2-丙烯酰胺基十二烷基磺酸共聚而成,丙烯酰胺、2-丙烯酰胺基-2-甲基丙磺酸和2-丙烯酰胺基十二烷基磺酸摩尔比优选为1:(0.1~40):(0.001~0.05),更优选为1∶(0.1~20)∶(0.001~0.01)。
上述技术方案中,所述耐温抗盐改性聚丙烯酰胺优选由丙烯酰胺、2-丙烯酰胺基-2-甲基丙磺酸共聚而成,丙烯酰胺与2-丙烯酰胺基-2-甲基丙磺酸摩尔比优选为(0.1~40)∶1,改性聚丙烯酰胺的黏均分子量优选为800~2500万。
上述技术方案中,所述碱为无机碱性物质或有机碱。
上述技术方案中,所述无机碱性物质优选碱金属氢氧化物、碱土金属氢氧化物、碱金属碳酸盐中的至少一种;进一步优选碱金属氢氧化物选自氢氧化钠、氢氧化钾中的至少一种,碱土金属氢氧化物选自氢氧化镁、氢氧化钙中的至少一种,碱金属碳酸盐选自碳酸钠或碳酸氢钠中的至少一种。
上述技术方案中,所述有机碱优选分子中含有伯胺基、仲胺基、叔胺基、季铵碱基中的至少一种,进一步优选为c1~c8短碳链有机胺中的至少一种,更优选为乙醇胺、二乙醇胺、三乙醇胺或三乙胺中的至少一种。
上述技术方案中,所述驱油剂中表面活性剂组合物与聚合物与碱的质量比优选为1∶(0.1~2):(0~5)。
本发明驱油剂组合物的关键有效成分是所述组分1)、2)和3),本领域技术人员知道,为了便于运输和贮存或现场使用等方面考虑,可以采用各种供应形式,例如不含水的固态形式,或者含水的固态形式,或者含水的膏状形式,或者水溶液形式;水溶液形式包括用水配成浓缩液的形式,直接配成现场驱油所需浓度的驱油剂形式;其中,对水没有特殊要求,可以是去离子水,还可以是含无机矿物质的水,而含无机矿物质的水可以是自来水、油田地层水或油田注入水。
本发明驱油剂组合物还可以含有本领域常用的泡沫剂、小分子有机物(例如乙二醇单甲醚、乙二醇单丁醚、dmso等)等采油助剂。
上述技术方案中,所述驱油剂可以采用各种常规混合方法按照所需量各组分混合得到,用于驱油时按照所需浓度用水溶解得到驱油剂用于驱油;还可以根据所需驱油剂的浓度,将所述驱油组合物中各组分分别溶解于水中得到驱油剂用于驱油。制备中所用的水可以为自来水、河水、海水,油田地层水。
上述技术方案中,所述驱油剂可以根据现有技术加以应用,可以单独使用,也可以与油田常用助剂复配使用;作为优选方案:所述应用优选油藏的地层盐水的总矿化度5000~200000mg/l,其中ca2++mg2+为10~15000mg/l、hco3-为0~2000mg/l;原油黏度为1.0~200.0mpa.s;地层温度为50~120℃。
为了解决上述技术问题之六,本发明采用的技术方案为:一种上述解决技术问题之五所述技术方案中任一所述的驱油剂的制备方法,包括以下步骤:
(a)季铵盐表面活性剂的制备:
将
(b)表面活性剂组合物的制备:
①在碱性催化剂存在下,r5nh2与所需量环氧乙烷、环氧丙烷、环氧乙烷反应得到聚醚化合物;将所需量的聚醚化合物与步骤(a)得到的季铵盐表面活性剂水溶液或小分子醇水溶液混合,得到所述的表面活性剂组合物;
或通过②进一步反应得到所述表面活性剂组合物:
②将步骤①得到的聚醚化合物与离子化试剂y0r6z或y0r'6z'以及碱金属氢氧化物或碱金属醇盐以摩尔比1:(0.1~20):(0.1~20)混合,搅拌下于反应温度50~120℃反应3~15小时,按所需摩尔比加入步骤(a)得到的季铵盐表面活性剂水溶液或小分子醇水溶液,升温至40~100℃搅拌1~5小时,得到所需的表面活性剂组合物驱油剂;其中,y0选自氯、溴或碘;
或:②将步骤①得到的聚醚化合物与离子化试剂y0r6coor0或y0r'6coor0以及碱金属氢氧化物或碱金属醇盐以摩尔比1:(0.1~20):(0.1~20)混合,搅拌下于反应温度50~120℃反应3~15小时,继续加入水进行皂化反应,回流1~10小时后,按所需摩尔比加入步骤(a)得到的季铵盐表面活性剂水溶液或小分子醇水溶液,升温至40~100℃搅拌1~5小时,得到所需的表面活性剂组合物;其中,y0选自氯、溴或碘,r0选自c1~c8的烷基。
(c)按所述质量份数计,将所需量的步骤(b)得到的表面活性剂组合物与聚合物、碱混合均匀,得到所述的驱油剂。
作为优选方案,其中,所述的(a)步骤中:所述压力优选为0.1~0.6mpa,小分子醇优选选自c1~c4的脂肪醇;所述的(b)步骤中:①所述的反应温度优选为120~160℃,压力优选为0.3~0.6mpa表压,碱性催化剂优选为氢氧化钾或无水碳酸钾中的至少一种;②所述碱金属氢氧化物优选为氢氧化钾或氢氧化钠中的至少一种,聚醚化合物与离子化试剂以及碱金属氢氧化物或碱金属醇盐的摩尔比优选为1:(2~10):(2~10),y0优选为氯或溴中的一种,r0优选为c1~c4的烷基。
本发明采用物理模拟驱替评价方法进行效果评价,评价方法为:将岩心恒温烘干至恒重,测定岩心的气测渗透率;以上述模拟油田地层水饱和岩心,计算其孔隙体积,于驱油温度下,以原油饱和岩心,记录饱和原油的体积,再以0.2ml/min的速度泵入地层水,驱至含水达100%,计算水驱提高原油的采收率,然后以0.15ml/min的速度转注0.1~1pv(岩心孔隙体积)步骤(c)得到的驱油剂,以0.2ml/min的速度水驱至含水100%,计算在水驱基础上提高原油采收率的百分数。
本发明界面张力的测试方法为:(1)预置温度至测定所需的温度,等待温度稳定;(2)注入外相液体,装满离心管,再注入内相液体,去除起泡,盖紧;(3)将离心管装入仪器的旋转轴内,设定转速,调节显微镜使视野中的内相液滴或气泡十分清晰;(4)读数与计算,按公式(1)计算界面张力:
γ=0.25ω2r3δρ(l/d≧4)公式(1);
其中,γ为界面张力(mn·m-1),δρ为两相密度差(kg·m-3.),ω为角速度(rad·s-1),r为液滴短轴半径(m)),l为长轴(离心管轴向)直径,d为短轴(离心管径向)直径。
本发明静态吸附量的测试方法为:表面活性剂的模拟盐水溶液与吸附质按一定液固比充分混合后,于设定温度和频率下震荡一定时间,冷却后离心分离,取上层清液,测定表面活性剂有效组分的浓度,计算表面活性剂吸附量,见公式(2);
γ=w(co×a-ce)/m公式(2);
其中,γ为静态吸附量(mg/g),w为表面活性剂溶液的重量(g),co为表面活性剂溶液的初始浓度(mg/g),a为表面活性剂产品的有效物含量(%),ce表面活性剂溶液的吸附后有效浓度(mg/g),m为吸附剂的质量(g)。
本发明增溶参数的测试方法为:(1)先将5ml耐温玻璃移液管的尖端封住,截取需用长度备用;(2)配制一定浓度的表面活性剂溶液,用移液器量取一定体积水溶液加入到尖端封口的玻璃移液管中,同时分析天平记录加入溶液的质量,按照同样的方法加入一定量的原油或者模拟油(油水比根据实验需要确定),记录体积和质量,记录水相和油相刻度;(3)样品加完后,密封玻璃移液管上口;(4)采用涡流震荡或旋转混合均匀;(5)在设定温度下静置一段时间,不断摇晃使其逐步达到平衡,拍照记录相态随时间的变化,计算增溶参数,见公式(3);
其中,sp*为增溶参数,vs、vo、vw分别为表面活性剂的体积、表面活性剂增溶原油的体积、表面活性剂增溶水的体积。
本发明羧化度和磺化度的测试方法为:用电位测量装置指示滴定分析过程中电位差(或电极电位)的变化来确定滴定终点的分析方法。利用电极的电极电位与待测组分的活度之间的关系来进行测定。
海尔敏阳离子溶液作滴定剂
s-+hyamine=s-hyamine
在碱性条件下(ph=11),羧酸盐和磺酸盐两种表面活性剂均以盐的形式存在,都能与海尔敏阳离子反应,用海尔敏阳离子溶液作滴定剂,可测得两种表面活性剂的含量。采用两相电位滴定法,以海尔敏阳离子溶液作滴定剂,通过电位滴定仪判别等当量电位可测得阴离子表面活性剂的羧化度或磺化度。
准确称取待测表面活性剂样品溶液5.0g,每次平行取样3-4份,记录称量重量ws(g),分别加入40ml蒸馏水,以0.2mnaoh标准溶液调节各平行样品ph值在11.00左右;在调好ph值的溶液中依次加入10ml乙醇、10ml甲基异丁基酮(mibk),采用0.004m的海明1622标准溶液滴定,记录消耗海尔敏的体积vh(ml)。采用下列公式计算表面活性剂样品的羧化度或磺化度。其中mw为待测表面活性剂样品的分子量。
采用本发明制备的表面活性剂组合物,以质量百分比计,用量为0.01~0.15wt%的范围内,可用于地层温度为50~120℃、矿化度为5000~200000毫克/升、mg2++ca2+为20~12000毫克/升、hco3-为0~2000mg/l的油田水和原油,测定了该表面活性剂水溶液与原油之间的动态界面张力值,可达10-2~10-4mn/m的低界面张力,静态吸附量小于2mg/g,4wt%表面活性剂能很好乳化原油,最大增溶参数为24.8,取得了较好的技术效果。
采用本发明的驱油剂,用于地层温度50~120℃、矿化度5000~200000毫克/升的模拟盐水和原油,以质量百分比计,用量为0.01~0.15wt%表面活性剂组合物与0~0.3wt%上述的聚合物与0~1.2wt%上述的碱形成驱油剂,测定了该驱油剂水溶液的表观黏度,与油田脱水原油之间的动态界面张力值可达10-2~10-4mn/m,静态吸附量小于2mg/g。经物理模拟驱替试验室内评价,该驱油剂能在水驱基础上提高原油采收率最高可达25.01%,取得了较好的技术效果。
附图说明
本发明制备的季铵盐表面活性剂可应用美国nicolet-5700光谱仪,采用全反射(atr)红外光谱法进行红外光谱分析(扫描范围4000~400cm-1),确定被测样品的化学结构,以达到对本发明所述化合物的红外表征。
图1为n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的红外光谱图。其中,3287.3cm-1为o-h伸缩振动特征峰,2930.3cm-1和2853.1m-1为甲基与亚甲基c-h伸缩特征峰,1508.6cm-1和1599.7cm-1为苯环的伸缩振动峰,1438.4cm-1为c-n弯曲振动吸收峰,1123.4cm-1和1228.4cm-1为c-n的伸缩振动峰,1053.5cm-1为伯醇中c-o的伸缩振动峰,、745.1cm-1和682.2cm-1为苯环中ch面外面内摇摆吸收峰。
图2为0.15%表面活性剂经不同时间老化后的油水界面张力图。
图3为室内岩心驱替实验流程图。
下面通过实施例对本发明作进一步阐述。
具体实施方式
【实施例1】
(a)n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的制备
将n,n-二羟乙基苯胺181.2克(1摩尔)与204.8克(1摩尔)1-氯十二烷、20wt%乙醇水溶液750克混合于配有机械搅拌、温度计和回流冷凝管的2000毫升的四口烧瓶内,加热至回流反应36小时,停止反应。取少量反应液进行lc-ms分析,n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的含量为98.5%,n,n-二羟乙基苯胺为0.7%,其余样品不处理,备用。
(b)表面活性剂组合物s01的制备
r5为ch3(ch2)7ch=ch(ch2)8;z1=z2=ch2coonar1+r2=3,s1+s2=12,r3+r4=4。
向装有搅拌装置的5l压力反应器中加入267.0克(1摩尔)9-烯-十八胺、9.5克氢氧化钾加热至80~90℃时,开启真空系统,在高真空下脱水1小时,然后用氮气置换3~4次,将体系反应温度调至110℃缓缓通入132.9克(3.02摩尔)环氧乙烷,控制压力≤0.50mpa,待环氧乙烷反应结束后,于150℃缓缓通入701.8克(12.1摩尔)环氧丙烷,控制压力≤0.60mpa,待环氧丙烷反应结束后再将温度调至130℃缓缓通入178.2克(4.05摩尔)环氧乙烷。反应结束后,降温至90℃,真空除去低沸物,冷却后中和、脱水,得9-烯十八胺聚氧乙烯(3)聚氧丙烯(12)聚氧乙烯(4)醚1230.3克,收率96.8%。
配有机械搅拌、温度计和回流冷凝管的5000毫升的反应瓶通氮气去除水气,氮气保护下加入9-烯-十八胺聚氧乙烯(3)聚氧丙烯(12)聚氧乙烯(4)醚635.5克(0.5摩尔)和108.0克(2.0摩尔)甲醇钠,缓慢滴入162.8克(1.5摩尔)氯乙酸甲酯,控制反应温度65℃反应8小时,冷却后加入1000克水及50克甲醇,继续加热至回流反应5小时。冷却,取50克均匀反应液以20wt%盐酸酸化,蒸除甲醇,加入100克苯混合,分去水层,以饱和食盐水洗涤3次,减压蒸去苯,得到的产物采用梅特勒公司t90自动电位滴定仪,以海尔敏阳离子溶液作滴定剂,测得羧化度为185.6%。剩余未处理的反应液加入1280克水、20.0克(0.5摩尔)氢氧化钠、含77.2克(0.2摩尔)n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s01。
【实施例2】
(a)n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的制备
将n,n-二羟乙基苯胺181.2克(1摩尔)与204.8克(1摩尔)1-氯十二烷、20wt%乙醇水溶液750克混合于配有机械搅拌、温度计和回流冷凝管的2000毫升的四口烧瓶内,加热至回流反应36小时,停止反应。取少量反应液进行lc-ms分析,n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的含量为98.5%,n,n-二羟乙基苯胺为0.7%,其余样品不处理,备用。
(b)表面活性剂组合物s02的制备
z1=z2=ch2cooh.hn(ch2ch2oh)2;r1+r2=4,s1+s2=15,r3+r4=3。
向装有搅拌装置的5l压力反应器中加入261克(1摩尔)十二烷基苯胺、5.9克氢氧化钠和11.9克无水碳酸钾,加热至80~90℃时,开启真空系统,在高真空下脱水1小时,然后用氮气置换3~4次,将体系反应温度调至110℃缓缓通入178.2克(4.05摩尔)环氧乙烷,控制压力≤0.50mpa,待环氧乙烷反应结束后,于150℃缓缓通入881.6克(15.2摩尔)环氧丙烷,控制压力≤0.60mpa,待环氧丙烷反应结束后再将温度调至140℃缓缓通入134.2克(3.05摩尔)环氧乙烷。反应结束后,降温至90℃,真空除去低沸物,冷却后中和、脱水,得十二烷基苯胺聚氧乙烯(4)聚氧丙烯(15)聚氧乙烯(3)醚1394.4克,收率96.9%。
十二烷基苯胺聚氧乙烯(4)聚氧丙烯(15)聚氧乙烯(3)醚719.5克(0.5摩尔)与60克(1.5摩尔)氢氧化钠、235.0克(2.0摩尔)氯乙酸钠及800毫升甲苯/苯(v/v=1)混合于配有机械搅拌、温度计和回流冷凝管的5000毫升的四口烧瓶内,升温至回流反应8小时。反应结束后将所有反应液进行酸化,以饱和食盐水洗涤3次,减压蒸去甲苯/苯,得到的产物采用梅特勒公司t90自动电位滴定仪,以海尔敏阳离子溶液作滴定剂,测得羧化度为190.4%,加入1500克水、126.0克(1.2摩尔)二乙醇胺、含38.6克(0.1摩尔)n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s02。
【实施例3】
(a)n,n,n-三甲基-(4-十二烷基)苯基氯化铵的制备
将n,n-二甲基-(4-十二烷基)苯胺289.0克(1摩尔)与50wt%异丙醇水溶液500克混合于2000毫升的压力釜中,经氮气多次置换去氧后,慢慢通入75.8克(1.5摩尔)氯甲烷,保持压力为0.3~0.5mpa反应6小时。降至常温,放空,抽去低沸物,取少量反应液进行lc-ms分析,n,n,n-三甲基-(4-十二烷基)苯基氯化铵的含量为96.8%,n,n-二甲基-(4-十二烷基)苯胺为1.2%。其余样品不处理,备用。
(b)表面活性剂组合物s03的制备
其中,r1+r2=5,s1+s2=5,r3+r4=3。
向装有搅拌装置的2l压力反应器中加入325克(1摩尔)二十二胺、2.3克氢氧化钾和8.4克无水碳酸钾,加热至80~90℃时,开启真空系统,在高真空下脱水1小时,然后用氮气置换3~4次,将体系反应温度调至120℃缓缓通入222.2克(5.05摩尔)环氧乙烷,控制压力≤0.60mpa,待环氧乙烷反应结束后再将温度调至130℃缓缓通入295.8克(5.1摩尔)环氧丙烷,控制压力≤0.60mpa,待环氧丙烷反应结束后再将温度调至140℃缓缓通入134.2克(3.05摩尔)环氧乙烷。反应结束后,降温至90℃,真空除去低沸物,冷却后中和、脱水,得二十二胺聚氧乙烯(5)聚氧丙烯(5)聚氧乙烯(3)醚925.4克,收率95.7%。
二十二胺聚氧乙烯(5)聚氧丙烯(5)聚氧乙烯(3)醚478.5克(0.5摩尔)与160克(4摩尔)氢氧化钠、589.5克(3摩尔)3-氯-2-羟基丙磺酸钠及800毫升甲苯/苯(v/v=2)混合于配有机械搅拌、温度计和回流冷凝管的5000毫升的四口烧瓶内,加热至85℃反应9小时。冷却,取50克均匀反应液以20wt%盐酸酸化,分去水及无机盐,蒸除溶剂,采用梅特勒公司t90自动电位滴定仪,以海尔敏阳离子溶液作滴定剂,通过电位滴测定磺化度为175.8%。剩余未处理的反应液蒸馏去除溶剂,加入600克水、80.0克(2.0摩尔)氢氧化钠、含74.7克(0.22摩尔)n,n,n-三甲基-(4-十二烷基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s03。
【实施例4】
(a)n,n,n-三甲基-(4-十二烷基)苯基氯化铵的制备
将n,n-二甲基-(4-十二烷基)苯胺289.0克(1摩尔)与50wt%异丙醇水溶液500克混合于2000毫升的压力釜中,经氮气多次置换去氧后,慢慢通入75.8克(1.5摩尔)氯甲烷,保持压力为0.3~0.5mpa反应6小时。降至常温,放空,抽去低沸物,取少量反应液进行lc-ms分析,n,n,n-三甲基-(4-十二烷基)苯基氯化铵的含量为96.8%,n,n-二甲基-(4-十二烷基)苯胺为1.2%。其余样品不处理,备用。
(b)表面活性剂组合物s04的制备
r5为ch3(ch2)7ch=ch(ch2)8;z1=z2=ch2coonar1+r2=3,s1+s2=12,r3+r4=4。
向装有搅拌装置的5l压力反应器中加入267.0克(1摩尔)9-烯-十八胺、9.5克氢氧化钾加热至80~90℃时,开启真空系统,在高真空下脱水1小时,然后用氮气置换3~4次,将体系反应温度调至110℃缓缓通入132.9克(3.02摩尔)环氧乙烷,控制压力≤0.50mpa,待环氧乙烷反应结束后,于150℃缓缓通入701.8克(12.1摩尔)环氧丙烷,控制压力≤0.60mpa,待环氧丙烷反应结束后再将温度调至130℃缓缓通入178.2克(4.05摩尔)环氧乙烷。反应结束后,降温至90℃,真空除去低沸物,冷却后中和、脱水,得9-烯十八胺聚氧乙烯(3)聚氧丙烯(12)聚氧乙烯(4)醚1230.3克,收率96.8%。
配有机械搅拌、温度计和回流冷凝管的5000毫升的反应瓶通氮气去除水气,氮气保护下加入9-烯-十八胺聚氧乙烯(3)聚氧丙烯(12)聚氧乙烯(4)醚635.5克(0.5摩尔)和108.0克(2.0摩尔)甲醇钠,缓慢滴入162.8克(1.5摩尔)氯乙酸甲酯,控制反应温度65℃反应8小时,冷却后加入1000克水及50克甲醇,继续加热至回流反应5小时。冷却,取50克均匀反应液以20wt%盐酸酸化,蒸除甲醇,加入100克苯混合,分去水层,以饱和食盐水洗涤3次,减压蒸去苯,得到的产物采用梅特勒公司t90自动电位滴定仪,以海尔敏阳离子溶液作滴定剂,测得羧化度为185.6%。剩余未处理的反应液蒸馏去除溶剂,加入1100克水、12.0克(0.3摩尔)氢氧化钠、含25.5克(0.075摩尔))n,n,n-三甲基-(4-十二烷基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s04。
【实施例5】
同【实施例3】,不同之处在于二十二胺聚氧乙烯(5)聚氧丙烯(5)聚氧乙烯(3)醚478.5克(0.5摩尔)无需继续反应,加入600克水,100克正丙醇,73.0克(1.0摩尔)正丁胺,8.5克(0.025摩尔)n,n,n-三甲基-(4-十二烷基)苯基氯化铵的异丙醇水溶液,得到所需的表面活性剂组合物s05。
【实施例6】
同【实施例1】,不同之处在于(a)步骤中反应结束时,减压蒸除乙醇,得到阳离子表面活性剂产品,其余相同,得到所需的表面活性剂组合物s06。
【实施例7】
(a)n,n,n-三甲基-(4-十六烷基)苯基氯化铵的制备
将n,n-二甲基-(4-十六烷基)苯胺345.0克(1摩尔)与80wt%异丙醇水溶液600克混合于2000毫升的压力釜中,经氮气多次置换去氧后,慢慢通入101.0克(2.0摩尔)氯甲烷,保持压力为0.3~0.5mpa反应8小时。降至常温,放空,抽去低沸物,取少量反应液进行lc-ms分析,n,n,n-三甲基-(4-十六烷基)苯基氯化铵的含量为95.4%,n,n-二甲基-(4-十六烷基)苯胺为1.9%。其余样品不处理,备用。
(b)表面活性剂组合物s07的制备
同【实施例3】得到剩余未处理的双磺酸盐反应液,蒸馏去除溶剂,加入600克水、80.0克(2.0摩尔)氢氧化钠、含59.3克(0.15摩尔)n,n,n-三甲基-(4-十六烷基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s07。
【实施例8】
(a)n-十二烷基-n,n-二乙基-(3-羟基)苯基溴化铵的制备
将n,n-二乙基-(3-羟基)苯胺165.0克(1摩尔)与249.0克(1摩尔)1-溴十二烷、50wt%异丙醇水溶液600克混合于配有机械搅拌、温度计和回流冷凝管的2000毫升的四口烧瓶内,加热至回流反应36小时,停止反应。取少量反应液进行lc-ms分析,n-十二烷基-n,n-二乙基-(3-羟基)苯基溴化铵的含量为98.8%,n,n-二乙基-(3-羟基)苯胺为0.3%,其余样品不处理,备用。
(b)表面活性剂组合物s08的制备
同【实施例1】得到剩余未处理的反应液,蒸馏去除溶剂,加入1780克水、20.0克(0.5摩尔)氢氧化钠、含372.6克(0.9摩尔)n-十二烷基-n,n-二乙基-(3-羟基)苯基溴化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s08。
【实施例9】
表面活性剂组合物作为驱油剂的性能实验。
配制不同盐含量的模拟水,组成见表1所示。实验用原油来自油田,原油性质见表2所示,经脱水后使用。
相态实验可以很好反映出表面活性剂对原油的增溶能力,得到表面活性剂对原油的增溶参数和表面活性剂的最佳盐含量。实验过程为:首先配制4.0wt%不同盐含量的水溶液(1#~9#模拟水),取2.5ml加入一端封口的5ml移液管中,再加入2.5ml脱水原油(油水体积比=1:1),上端分封口后,记录起始的油水体积,充分混合后,放入不锈钢密封容器中置于烘箱恒温静置,直到各相体积不变为至,记录各相体积,计算表面活性剂对原油的增溶参数,增溶参数最大时的盐度为最佳盐含量,结果见表2所示。
静态吸附试验主要是从研究表面活性剂在地层岩心上的吸附损耗量入手,探索实施例合成的表面活性剂在提高原油采收率现场应用的经济性和可形性。实验过程为:表面活性剂的模拟盐水溶液3g与1g含黏土的石英砂混合后,于设定温度下震荡24h,冷却后离心分离,取上层清液,采用toc方法测定表面活性剂有效组分的浓度,计算表面活性剂吸附量,单位mg/g,结果见表3所示。其中,含黏土的石英砂组成为:10wt%高岭土+90wt%100~200目石英砂。
将表面活性剂组合物以相应的模拟水溶解,测定表面活性剂溶液对原油的油水界面张力,结果见表4所示。将0.15wt%的表面活性剂组合物模拟盐水溶液装入20毫升安碚瓶中,密封后放入烘箱内,测定不同老化时间后的油水界面张力,发现老化后油水界面张力仍可保持10-3~10-4mn/m的超低值,见图2所示。油水界面张力(ift)由美国德克萨斯大学生产的tx500型旋转滴界面张力仪测定。
【比较例1】
同【实施例3】,不同之处在于步骤(a)中以“n,n-二甲基-(4-十二烷基)苄胺303.0克(1摩尔)”替代“n,n-二甲基-(4-十二烷基)苯胺289.0克(1摩尔)”,其余相同,得到表面活性剂组合物s09。同【实施例9】进行性能实验,结果见表5所示。
【比较例2】
同【实施例3】,不同之处在于以市售的苯基三甲基氯化铵、苄基三乙基氯化铵替代n,n,n-三甲基-(4-十二烷基)苯基氯化铵,其余相同,得到表面活性剂组合物s10、s11。同【实施例9】进行性能实验,结果见表5所示。
【比较例3】
同【实施例3】,不同之处在于分别以市售的n-十二烷基-n,n-二(2-羟乙基)苄基氯化铵、十二烷基二甲基苄基氯化铵、十八烷基二甲基苄基氯化铵替代n,n,n-三甲基-(4-十二烷基)苯基氯化铵,其余相同,得到表面活性剂组合物s12、s13和s14。同【实施例9】进行性能实验,结果见表5所示。
【比较例4】
c18h37(ch2ch2oh)2n+(ch2)4n+(ch2ch2oh)2c18h37.2br-(18-4-18,2br-)
同【实施例1】,不同之处在于以双子阳离子季铵盐表面活性剂(18-4-18,2br-)替代n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵,其余相同,得到表面活性剂组合物s15。同【实施例9】进行性能实验,结果见表5所示。
【比较例5】
同【实施例1】,不同之处在于,与环氧丙烷和环氧乙烷不是先后分步进行反应的,而是混合后一步进行反应,其余相同,得到s16,同【实施例9】进行性能实验,结果见表5所示。
表1
表2
表3
表4
表5
【实施例10】
(a)n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的制备
将n,n-二羟乙基苯胺181.2克(1摩尔)与204.8克(1摩尔)1-氯十二烷、20wt%乙醇水溶液750克混合于配有机械搅拌、温度计和回流冷凝管的2000毫升的四口烧瓶内,加热至回流反应36小时,停止反应。取少量反应液进行lc-ms分析,n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的含量为98.5%,n,n-二羟乙基苯胺为0.7%,其余样品不处理,备用。
(b)表面活性剂组合物s01的制备
r5为ch3(ch2)7ch=ch(ch2)8;z1=z2=ch2coonar1+r2=3,s1+s2=12,r3+r4=4。
向装有搅拌装置的5l压力反应器中加入267.0克(1摩尔)9-烯-十八胺、9.5克氢氧化钾加热至80~90℃时,开启真空系统,在高真空下脱水1小时,然后用氮气置换3~4次,将体系反应温度调至110℃缓缓通入132.9克(3.02摩尔)环氧乙烷,控制压力≤0.50mpa,待环氧乙烷反应结束后,于150℃缓缓通入701.8克(12.1摩尔)环氧丙烷,控制压力≤0.60mpa,待环氧丙烷反应结束后再将温度调至130℃缓缓通入178.2克(4.05摩尔)环氧乙烷。反应结束后,降温至90℃,真空除去低沸物,冷却后中和、脱水,得9-烯十八胺聚氧乙烯(3)聚氧丙烯(12)聚氧乙烯(4)醚1230.3克,收率96.8%。
配有机械搅拌、温度计和回流冷凝管的5000毫升的反应瓶通氮气去除水气,氮气保护下加入9-烯-十八胺聚氧乙烯(3)聚氧丙烯(12)聚氧乙烯(4)醚635.5克(0.5摩尔)和108.0克(2.0摩尔)甲醇钠,缓慢滴入162.8克(1.5摩尔)氯乙酸甲酯,控制反应温度65℃反应8小时,冷却后加入1000克水及50克甲醇,继续加热至回流反应5小时。冷却,取50克均匀反应液以20wt%盐酸酸化,蒸除甲醇,加入100克苯混合,分去水层,以饱和食盐水洗涤3次,减压蒸去苯,得到的产物采用梅特勒公司t90自动电位滴定仪,以海尔敏阳离子溶液作滴定剂,测得羧化度为185.6%。剩余未处理的反应液加入1280克水、20.0克(0.5摩尔)氢氧化钠、含77.2克(0.2摩尔)n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s01。
配制不同盐含量的模拟水,组成见表1所示。实验用原油来至油田,原油性质见表2所示,经脱水后使用。
相态实验可以很好反映出表面活性剂对原油的增溶能力,得到表面活性剂对原油的增溶参数和表面活性剂的最佳盐含量。实验过程为:首先配制4.0wt%不同盐含量的表面活性剂水溶液(1#~9#模拟水),取2.5ml加入一端封口的5ml移液管中,再加入2.5ml脱水原油(油水体积比=1:1),上端分封口后,记录起始的油水体积,充分混合后,放入不锈钢密封容器中置于烘箱恒温静置,直到各相体积不变为至,记录各相体积,计算表面活性剂对原油的增溶参数,增溶参数最大时的盐度为最佳盐含量,结果见表2所示。
将表面活性剂模拟水溶液3g与1g含黏土的石英砂混合后震荡24h,冷却后离心分离,取上层清液测定吸附量,单位mg/g,结果见表3所示。其中,吸附量采用toc方法测定,含黏土的石英砂组成为:10wt%高岭土+90wt%100~200目石英砂。
(c)驱油剂的性能实验
(1)驱油剂水溶液的配制
以模拟水配制s01表面活性剂组合物、改性聚丙烯酰胺聚合物(p1,共聚am/amps摩尔比=1/0.05,黏均分子量2500万)和二乙醇胺的水溶液,再按所需比例混合稀释得到均匀的驱油剂。
(2)测定驱油剂的黏度及油水界面张力,并与s01、p1相比较,见表6所示。其中,表观黏度由haakemarsiii型旋转流变仪测定,界面张力由美国德克萨斯大学生产的tx500型旋转滴界面张力仪测定。
(3)将人造岩心恒温烘干至恒重,测量岩心的平均直径及岩心长度,称量岩心干重,测定岩心的气测渗透率。以上述模拟盐水饱和岩心,测试其孔隙体积。以油田脱水原油饱和岩心,记录饱和原油的体积。于70℃温度下,以11#模拟水驱至采出液含水达100%,计算水驱提高原油的采收率,转注0.3pv(岩心孔隙体积)驱油剂后,水驱至含水100%,计算在水驱基础上提高原油采收率的百分数,同时与注相同pv的表面活性剂和聚合物相对比,见表6所示。采用的模拟岩心驱替试验流程如图3所示。脱水原油黏度为1.9mpa.s。
【实施例11】
(a)n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的制备
将n,n-二羟乙基苯胺181.2克(1摩尔)与204.8克(1摩尔)1-氯十二烷、20wt%乙醇水溶液750克混合于配有机械搅拌、温度计和回流冷凝管的2000毫升的四口烧瓶内,加热至回流反应36小时,停止反应。取少量反应液进行lc-ms分析,n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的含量为98.5%,n,n-二羟乙基苯胺为0.7%,其余样品不处理,备用。
(b)表面活性剂组合物s02的制备
z1=z2=ch2cooh.hn(ch2ch2oh)2;r1+r2=4,s1+s2=15,r3+r4=3。
向装有搅拌装置的5l压力反应器中加入261克(1摩尔)十二烷基苯胺、5.9克氢氧化钠和11.9克无水碳酸钾,加热至80~90℃时,开启真空系统,在高真空下脱水1小时,然后用氮气置换3~4次,将体系反应温度调至110℃缓缓通入178.2克(4.05摩尔)环氧乙烷,控制压力≤0.50mpa,待环氧乙烷反应结束后,于150℃缓缓通入881.6克(15.2摩尔)环氧丙烷,控制压力≤0.60mpa,待环氧丙烷反应结束后再将温度调至140℃缓缓通入134.2克(3.05摩尔)环氧乙烷。反应结束后,降温至90℃,真空除去低沸物,冷却后中和、脱水,得十二烷基苯胺聚氧乙烯(4)聚氧丙烯(15)聚氧乙烯(3)醚1394.4克,收率96.9%。
十二烷基苯胺聚氧乙烯(4)聚氧丙烯(15)聚氧乙烯(3)醚719.5克(0.5摩尔)与60克(1.5摩尔)氢氧化钠、235.0克(2.0摩尔)氯乙酸钠及800毫升甲苯/苯(v/v=1)混合于配有机械搅拌、温度计和回流冷凝管的5000毫升的四口烧瓶内,升温至回流反应8小时。反应结束后将所有反应液进行酸化,以饱和食盐水洗涤3次,减压蒸去甲苯/苯,得到的产物采用梅特勒公司t90自动电位滴定仪,以海尔敏阳离子溶液作滴定剂,测得羧化度为190.4%,加入1500克水、126.0克(1.2摩尔)二乙醇胺、含38.6克(0.1摩尔)n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s02。
同【实施例10】进行相态和静态吸附实验,结果见表2和表3所示。
(c)驱油剂的性能实验
同【实施例10】,不同之处在于以s02替代s01,以碳酸钾替代二乙醇胺,温度为65℃,脱水原油黏度为122.6mpa.s,结果见表6所示。
【实施例12】
(a)n,n,n-三甲基-(4-十二烷基)苯基氯化铵的制备
将n,n-二甲基-(4-十二烷基)苯胺289.0克(1摩尔)与50wt%异丙醇水溶液500克混合于2000毫升的压力釜中,经氮气多次置换去氧后,慢慢通入75.8克(1.5摩尔)氯甲烷,保持压力为0.3~0.5mpa反应6小时。降至常温,放空,抽去低沸物,取少量反应液进行lc-ms分析,n,n,n-三甲基-(4-十二烷基)苯基氯化铵的含量为96.8%,n,n-二甲基-(4-十二烷基)苯胺为1.2%。其余样品不处理,备用。
(b)表面活性剂组合物s03的制备
其中,r1+r2=5,s1+s2=5,r3+r4=3。
向装有搅拌装置的2l压力反应器中加入325克(1摩尔)二十二胺、2.3克氢氧化钾和8.4克无水碳酸钾,加热至80~90℃时,开启真空系统,在高真空下脱水1小时,然后用氮气置换3~4次,将体系反应温度调至120℃缓缓通入222.2克(5.05摩尔)环氧乙烷,控制压力≤0.60mpa,待环氧乙烷反应结束后再将温度调至130℃缓缓通入295.8克(5.1摩尔)环氧丙烷,控制压力≤0.60mpa,待环氧丙烷反应结束后再将温度调至140℃缓缓通入134.2克(3.05摩尔)环氧乙烷。反应结束后,降温至90℃,真空除去低沸物,冷却后中和、脱水,得二十二胺聚氧乙烯(5)聚氧丙烯(5)聚氧乙烯(3)醚925.4克,收率95.7%。
二十二胺聚氧乙烯(5)聚氧丙烯(5)聚氧乙烯(3)醚478.5克(0.5摩尔)与160克(4摩尔)氢氧化钠、589.5克(3摩尔)3-氯-2-羟基丙磺酸钠及800毫升甲苯/苯(v/v=2)混合于配有机械搅拌、温度计和回流冷凝管的5000毫升的四口烧瓶内,加热至85℃反应9小时。冷却,取50克均匀反应液以20wt%盐酸酸化,分去水及无机盐,蒸除溶剂,采用梅特勒公司t90自动电位滴定仪,以海尔敏阳离子溶液作滴定剂,通过电位滴测定磺化度为175.8%。剩余未处理的反应液蒸馏去除溶剂,加入600克水、80.0克(2.0摩尔)氢氧化钠、含74.7克(0.22摩尔)n,n,n-三甲基-(4-十二烷基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s03。
同【实施例10】进行相态和静态吸附实验,结果见表2和表3所示。
(c)驱油剂的性能实验
同【实施例10】,不同之处在于以s03替代s01、以疏水缔合聚合物p2(共聚am/amps/2-丙烯酰胺基十二烷基磺酸摩尔比=1/0.45/0.002,黏均分子量1750万)替代p1、以乙醇胺替代二乙醇胺配制驱油剂水溶液,水为12#模拟水,温度为98℃,脱水原油黏度为3.2mpa.s,结果见表7所示。
【实施例13】
(a)n,n,n-三甲基-(4-十二烷基)苯基氯化铵的制备
将n,n-二甲基-(4-十二烷基)苯胺289.0克(1摩尔)与50wt%异丙醇水溶液500克混合于2000毫升的压力釜中,经氮气多次置换去氧后,慢慢通入75.8克(1.5摩尔)氯甲烷,保持压力为0.3~0.5mpa反应6小时。降至常温,放空,抽去低沸物,取少量反应液进行lc-ms分析,n,n,n-三甲基-(4-十二烷基)苯基氯化铵的含量为96.8%,n,n-二甲基-(4-十二烷基)苯胺为1.2%。其余样品不处理,备用。
(b)表面活性剂组合物s04的制备
r5为ch3(ch2)7ch=ch(ch2)8;z1=z2=ch2coonar1+r2=3,s1+s2=12,r3+r4=4。
向装有搅拌装置的5l压力反应器中加入267.0克(1摩尔)9-烯-十八胺、9.5克氢氧化钾加热至80~90℃时,开启真空系统,在高真空下脱水1小时,然后用氮气置换3~4次,将体系反应温度调至110℃缓缓通入132.9克(3.02摩尔)环氧乙烷,控制压力≤0.50mpa,待环氧乙烷反应结束后,于150℃缓缓通入701.8克(12.1摩尔)环氧丙烷,控制压力≤0.60mpa,待环氧丙烷反应结束后再将温度调至130℃缓缓通入178.2克(4.05摩尔)环氧乙烷。反应结束后,降温至90℃,真空除去低沸物,冷却后中和、脱水,得9-烯十八胺聚氧乙烯(3)聚氧丙烯(12)聚氧乙烯(4)醚1230.3克,收率96.8%。
配有机械搅拌、温度计和回流冷凝管的5000毫升的反应瓶通氮气去除水气,氮气保护下加入9-烯-十八胺聚氧乙烯(3)聚氧丙烯(12)聚氧乙烯(4)醚635.5克(0.5摩尔)和108.0克(2.0摩尔)甲醇钠,缓慢滴入162.8克(1.5摩尔)氯乙酸甲酯,控制反应温度65℃反应8小时,冷却后加入1000克水及50克甲醇,继续加热至回流反应5小时。冷却,取50克均匀反应液以20wt%盐酸酸化,蒸除甲醇,加入100克苯混合,分去水层,以饱和食盐水洗涤3次,减压蒸去苯,得到的产物采用梅特勒公司t90自动电位滴定仪,以海尔敏阳离子溶液作滴定剂,测得羧化度为185.6%,红外分析图谱见图1所示。剩余未处理的反应液蒸馏去除溶剂,加入1100克水、12.0克(0.3摩尔)氢氧化钠、含25.5克(0.075摩尔))n,n,n-三甲基-(4-十二烷基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s04。
同【实施例10】进行相态和静态吸附实验,结果见表2和表3所示。
(c)驱油剂的性能实验
同【实施例10】,不同之处在于以s04替代s01,以碳酸钠替代二乙醇胺,结果见表6所示。
【实施例14】
同【实施例3】,不同之处在于二十二胺聚氧乙烯(5)聚氧丙烯(5)聚氧乙烯(3)醚478.5克(0.5摩尔)无需继续反应,加入600克水,100克正丙醇,73.0克(1.0摩尔)正丁胺,8.5克(0.025摩尔)n,n,n-三甲基-(4-十二烷基)苯基氯化铵的异丙醇水溶液,得到所需的表面活性剂组合物s05。
同【实施例10】进行相态和静态吸附实验,结果见表2和表3所示。
以s07替代s03配制驱油剂水溶液,同【实施例12】进行驱油实验,结果见表7所示。
【实施例15】
同【实施例1】,不同之处在于(a)步骤中反应结束时,减压蒸除乙醇,得到阳离子表面活性剂产品,其余相同,得到所需的表面活性剂组合物s06。同【实施例10】进行相态和静态吸附实验,结果见表2和表3所示。同【实施例10】进行驱油实验,结果见表6所示。
【实施例16】
(a)n,n,n-三甲基-(4-十六烷基)苯基氯化铵的制备
将n,n-二甲基-(4-十六烷基)苯胺345.0克(1摩尔)与80wt%异丙醇水溶液600克混合于2000毫升的压力釜中,经氮气多次置换去氧后,慢慢通入101.0克(2.0摩尔)氯甲烷,保持压力为0.3~0.5mpa反应8小时。降至常温,放空,抽去低沸物,取少量反应液进行lc-ms分析,n,n,n-三甲基-(4-十六烷基)苯基氯化铵的含量为95.4%,n,n-二甲基-(4-十六烷基)苯胺为1.9%。其余样品不处理,备用。
(b)表面活性剂组合物s07的制备
同【实施例3】得到剩余未处理的双磺酸盐反应液,蒸馏去除溶剂,加入600克水、80.0克(2.0摩尔)氢氧化钠、含59.3克(0.15摩尔)n,n,n-三甲基-(4-十六烷基)苯基氯化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s07。同【实施例10】进行相态和静态吸附实验,结果见表2和表3所示。同【实施例12】进行驱油实验,结果见表7所示。
【实施例17】
(a)n-十二烷基-n,n-二乙基-(3-羟基)苯基溴化铵的制备
将n,n-二乙基-(3-羟基)苯胺165.0克(1摩尔)与249.0克(1摩尔)1-溴十二烷、50wt%异丙醇水溶液600克混合于配有机械搅拌、温度计和回流冷凝管的2000毫升的四口烧瓶内,加热至回流反应36小时,停止反应。取少量反应液进行lc-ms分析,n-十二烷基-n,n-二乙基-(3-羟基)苯基溴化铵的含量为98.8%,n,n-二乙基-(3-羟基)苯胺为0.3%,其余样品不处理,备用。
(b)表面活性剂组合物s08的制备
同【实施例1】得到剩余未处理的反应液,蒸馏去除溶剂,加入1780克水、20.0克(0.5摩尔)氢氧化钠、含372.6克(0.9摩尔)n-十二烷基-n,n-二乙基-(3-羟基)苯基溴化铵的乙醇水溶液,混合均匀得到所需的表面活性剂组合物s08。性能结果见表2、表3和表6所示。
【比较例6】
同【实施例12】,不同之处在于步骤(a)中以n,n-二甲基-(4-十二烷基)苄胺303.0克(1摩尔)替代n,n-二甲基-(4-十二烷基)苯胺289.0克(1摩尔),其余相同,得到表面活性剂组合物s09,结果见表5和表8所示。
【比较例7】
同【实施12】,不同之处在于以市售的苯基三甲基氯化铵、苄基三乙基氯化铵替代n,n,n-三甲基-(4-十二烷基)苯基氯化铵,其余相同,得到表面活性剂组合物s10、s11,结果见表5和表8所示。
【比较例8】
同【实施例12】,不同之处在于分别以市售的n-十二烷基-n,n-二(2-羟乙基)苄基氯化铵、十二烷基二甲基苄基氯化铵、十八烷基二甲基苄基氯化铵替代n,n,n-三甲基-(4-十二烷基)苯基氯化铵,其余相同,得到表面活性剂组合物s12、s13和s14,结果见表5和表8所示。
【比较例9】
c18h37(ch2ch2oh)2n+(ch2)4n+(ch2ch2oh)2c18h37.2br-(18-4-18,2br-)
同【实施例10】,不同之处在于以双子阳离子季铵盐表面活性剂(18-4-18,2br-)替代n-十二烷基-n,n-二(2-羟乙基)苯基氯化铵,其余相同,得到表面活性剂组合物s15,结果见5和表8所示。
【比较例10】
同【实施例10】,不同之处在于,与环氧丙烷和环氧乙烷不是先后分步进行反应的,而是混合后一步进行反应,其余相同,得到s16,结果见表5和表8所示。
【比较例11】
同【实施例10】,不同之处在于,以高分子量阴离子聚丙烯酰胺p3(黏均分子量为2300万)替代改性缔合聚合物p1,其余相同,结果见8所示。
表6
表7
表8
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



