
 商标分类
商标分类  商标转让
商标转让 
反应性分散染料及其制备方法和染色应用与流程
 2021-02-02 15:02:19|
2021-02-02 15:02:19| 282|
282| 起点商标网
起点商标网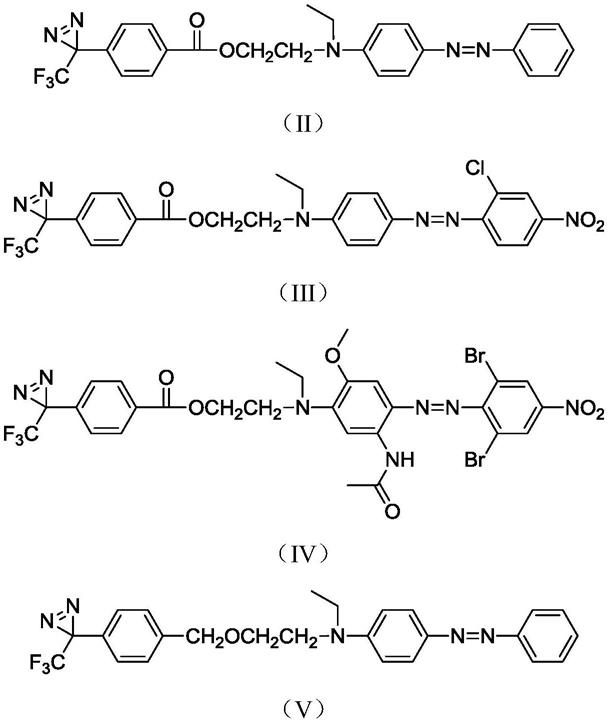
[0001]
本发明属于精细化工领域及纺织印染领域,涉及一种反应性分散染料、其制备方法及染色应用。
背景技术:
[0002]
涤纶织物通常以分散染料进行染色,分散染料与涤纶纤维之间主要以范德华力、氢键、疏水作用等弱作用力结合,因此,分散染料分子在高湿热等严苛条件下仍然会离开涤纶纤维,导致染色涤纶织物产生色牢度下降等问题。
[0003]
现有反应性分散染料,亦称活性分散染料或分散活性染料,通常是指以分散染料为母体,引入卤代三嗪基、β-羟乙基砜硫酸酯基等反应性基团,而生成的一类染料,专用于涤棉、涤毛等混纺织物染色。该类染料兼具分散染料的疏水性及活性染料的反应性,因而可同时与混纺织物中的合成纤维和天然纤维着色(参考:李云祥,青岛化工,1989,(1),21-23)。
[0004]
但是现有反应性分散染料由于活性基团的反应能力不足,仅能与棉、毛等天然纤维反应,仍然无法对涤纶纤维进行反应性着色。
[0005]
双吖丙啶类化合物是一类新兴物质,在材料、化学、生物等领域中受到广泛研究。据报道(参考:lepage et al.,science 2019,366,875-878),双吖丙啶结构在高温(>100℃)或紫外光照射(波长350nm左右)条件下,会形成活泼的卡宾中间体,从而具有插入氧-氢键、氮-氢键甚至碳-氢键的能力。双吖丙啶结构通式如下:
[0006][0007]
尚未见以双吖丙啶结构为反应性基团的反应性分散染料。
技术实现要素:
[0008]
本发明要解决的问题是提供一种反应性分散染料、其制备方法及其染色应用。
[0009]
为了解决上述技术问题,本发明提供一种反应性分散染料,其具有如下结构通式(i):
[0010][0011][0012]
其中,
[0013]
x为-cooch
2
ch
2-、-ch
2
och
2
ch
2-、-co-或-ch
2-;
[0014]
r
1
为氢原子、甲基、乙基、正丙基、异丙基、正丁基、正戊基、正己基、环己基、苯甲基或氰乙基;
[0015]
r
2
、r
3
分别为氢原子、甲基、乙基、甲氧基、乙氧基、羟基、甲氨基、二甲氨基、乙酰氨基或环己基氨基;
[0016]
r
4
、r
5
、r
6
分别选自氢原子、硝基、氯原子、溴原子、氰基、乙酰基或苯甲酰基。
[0017]
作为本发明的反应性分散染料的改进,结构式为以下任一:
[0018]
[0019][0020]
本发明还同时提供了上述反应性分散染料的制备方法,为以下任一方法:
[0021]
方法一:
[0022]
氮气保护下,将原料a、原料c、二环己基碳二亚胺、4-二甲氨基吡啶、二氯甲烷加入
至容器(例如为三口烧瓶)中,搅拌反应;反应温度为20~30℃,所述反应时间为12
±
1h;所述原料a、原料c、二环己基碳二亚胺、4-二甲氨基吡啶的摩尔比为1:1:1:0.1;每0.05~1.0mol的原料a配用1l的二氯甲烷;反应液经后处理(包括硅胶柱分离等),得反应性分散染料化合物;
[0023]
方法二、
[0024]
氮气保护下,将原料c、氢化钠、四氢呋喃加入至容器(例如为三口烧瓶)中,搅拌反应;反应温度为0~5℃,反应时间为30
±
5min;所述原料c、氢化钠的摩尔比为1:1,每0.05~1.0mol的原料c配用1l的四氢呋喃;然后加入原料b,继续搅拌反应;反应温度为20~30℃,反应时间为12
±
1h;所述原料b与原料c的摩尔比为1:1;反应液经后处理(包括硅胶柱分离等),得反应性分散染料化合物;
[0025]
原料a的结构为:
[0026][0027]
原料b的结构为:
[0028][0029]
原料c的结构为:
[0030][0031]
其中,
[0032]
x为氢原子或hoch
2
ch
2-;
[0033]
r
1
为氢原子、甲基、乙基、正丙基、异丙基、正丁基、正戊基、正己基、环己基、苯甲基或氰乙基;
[0034]
r
2
、r
3
为选自氢原子、甲基、乙基、甲氧基、乙氧基、羟基、甲氨基、二甲氨基、乙酰氨基或环己基氨基;
[0035]
r
4
、r
5
、r
6
为选自氢原子、硝基、氯原子、溴原子、氰基、乙酰基或苯甲酰基。
[0036]
本发明还同时提供了利用上述反应性分散染料制备而得的分散染料混合物:所述分散染料混合物由反应性分散染料和分散剂按照1:0.5~2的质量比组成;
[0037]
所述分散剂为分散剂nno、分散剂mf及木质素磺酸钠。
[0038]
本发明还同时提供了上述分散染料混合物的制备方法,包括以下步骤:
[0039]
(1)将反应性分散染料、分散剂、水混合后进行高速分散砂磨(于砂磨机中),砂磨速度为3000
±
300转/分,砂磨时间2
±
0.5小时,得分散液;所述水与反应性分散染料的质量
比为9~11:1,所述反应性分散染料与分散剂的质量比为1:0.5~2;
[0040]
(2)将步骤(1)所得的分散液进行喷雾干燥,雾化速度为15~20ml/min,进口温度300
±
30℃,出口温度100
±
10℃,得到分散染料混合物。
[0041]
本发明还同时提供了上述分散染料混合物进行的染色方法,包括如下步骤:
[0042]
(1)在染缸中,将0.5~5%owf的分散染料混合物配制成水溶液,调节ph至5(采用醋酸),加入5%owf的促染剂;所述促染剂为丁二酸二丁酯;
[0043]
(2)放入涤纶织物至染缸中,密封染缸,升温至保温温度;所述升温速率为2℃/min,所述保温温度为95
±
2℃;所述保温时间为30~60min(优选30min);
[0044]
(3)冷却至室温后,取出织物,用清水冲洗,晾干后,焙烘,即得染色涤纶织物;所述焙烘温度为120~180℃,优选140~180℃,更优选160℃;所述焙烘时间为0.5~2h,更优选2h。
[0045]
在发明过程中,发明人设想将双吖丙啶结构用于染料领域,将双吖丙啶结构作为反应性基团与疏水性染料母体结合,从而创造一类新的反应性分散染料,有可能使染料分子能够与涤纶纤维间形成共价键结合。然而实际应用时发现,合成所得的双吖丙啶染料在使用常规涤纶织物染色法时,所得染色织物仅有很少量的染料能与涤纶纤维以共价键结合,固色率低下。
[0046]
双吖丙啶结构应用于染料领域的问题在于:(1)卡宾中间体过于活泼,无法控制其仅和涤纶纤维反应;(2)涤纶染色通常需要高温,按文献推断,双吖丙啶必定会转化成卡宾;(3)染色机理及染色过程复杂等。
[0047]
具体来说,涤纶纤维,即聚对苯二甲酸乙二醇酯,结构中无活泼氢,仅有脂肪族碳-氢键和苯环上碳-氢键,反应优先级较低;而染色时环境复杂,通常需要添加分散剂、匀染剂等助剂,染色的过程是染料首先在水相中解聚成单分子染料,随后扩散至纤维表面,进而进入纤维内部,在染色高温条件下,双吖丙啶染料不可避免地会在水相中生成卡宾并优先与水分子发生反应而失活,在纤维表面残留的大量助剂亦有竞争反应的可能。
[0048]
另一方面,为获得广泛的色谱,染料发色体结构中通常会包含羟基、氨基等基团,亦会参与与卡宾的竞争反应,不利于染料与纤维间的成键反应。
[0049]
因此,本发明设定了特定的染色方法,从而解决上述技术问题。
[0050]
本发明与现有技术相比,具有以下技术效果:
[0051]
本发明的反应性分散染料可与涤纶纤维发生化学反应,形成共价键而牢固结合;本发明特定的染料分子结构,限定了染料分子结构中氧-氢键、氮-氢键的数量,能有效防止卡宾中间体与染料本身的副反应;染色时采用先低温(<100℃)染色后高温反应固色的方法,意外地发现双吖丙啶染料在约95℃、30min的染色条件下,能有效避免染料失活,得到高反应固色率;与常规分散染料相比,所染涤纶织物具有更好的耐皂洗、耐摩擦、耐升华及耐溶剂萃取等色牢度;染料合成方便、染色固色操作简单、固色仅需高温处理,具有广阔的应用前景。
附图说明
[0052]
下面结合附图对本发明的具体实施方式作进一步详细说明。
[0053]
图1染料(ii)的核磁共振氢谱;
[0054]
图2染料(ii)的核磁共振碳谱;
[0055]
图3染料(ii)在n,n-二甲基甲酰胺(dmf)中的紫外可见吸收光谱;
[0056]
图4染料(ii)染色涤纶织物的k/s曲线;
[0057]
图5染料(iii)的核磁共振氢谱;
[0058]
图6染料(iii)的核磁共振碳谱;
[0059]
图7染料(iii)在n,n-二甲基甲酰胺(dmf)中的紫外可见吸收光谱;
[0060]
图8染料(iii)染色涤纶织物的k/s曲线;
[0061]
图9反应性分散染料与涤纶纤维的反应机理图。
具体实施方式
[0062]
下面通过具体实施例对本发明进行详细和具体地介绍,以使更好地理解本发明,但是下述实施例并不限制本发明范围。化合物1、化合物5和化合物12可由市售获得,亦可按照文献(j.fluorine chem.2012,140,62-69;angew.chem.int.ed.2018,57,16688-16692;proceedings of the national academy of sciences of belarus,chemical series,2019,55(2),171-174)提供方法合成获得;化合物2-4、6-11可由市售获得。
[0063]
实施例1:染料(ii)的合成
[0064][0065]
在氮气保护、室温条件下,依次向三口瓶中加入化合物1(2mmol,0.460g)、化合物2(2mmol,0.534g)、二环己基碳二亚胺(2mmol,0.413g)、4-二甲氨基吡啶(0.2mmol,0.025g)和二氯甲烷(20ml),于20~30℃搅拌反应12h,采用薄层色谱法监测反应,12h后原料已经消耗完毕停止反应。
[0066]
加40ml水、20ml二氯甲烷,萃取三次(二氯甲烷40ml
×
3),合并有机相,使用旋转蒸发仪蒸除溶剂(二氯甲烷),残留物经200~300目的硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯=5/1,v/v)得到染料(ii),黄色固体,0.80g,收率83.1%。
[0067][0068]
1
h nmr(cdcl
3
,400mhz)δ8.03(d,j=7.6hz,2h),7.88(d,j=7.6hz,2h),7.85(d,j=7.6hz,2h),7.48(dd,j
1
=j
2
=7.6hz,2h),7.39(dd,j
1
=7.2hz,j
2
=7.6hz,1h),7.24(d,j=7.6hz,2h),6.84(d,j=7.6hz,2h),4.54(t,j
1
=6.4hz,j
2
=6.0hz,2h),3.80(t,j
1
=6.4hz,j
2
=6.0hz,2h),3.55(q,j=7.2hz,2h),1.26(t,j=7.2hz,3h).
13
c nmr(cdcl
3
,100mhz)δ165.52,153.19,150.02,143.90,134.06,130.87,129.98,129.52,128.96,126.45,125.22,122.25,121.86(q,j=273hz),111.52,62.53,48.82,45.60,29.72,12.36.esi-ms:m/z=482.2[m+h].
[0069]
实施例2:染料(iii)的合成
[0070]
将化合物2替换为化合物3(2mmol,0.697g),其余等同于实施例1,得到染料(iii),红色固体,0.98g,收率87.4%。
[0071][0072]
1
h nmr(cdcl
3
,400mhz)δ8.40(d,j=2.4hz,1h),8.16(dd,j
1
=8.4hz,j
2
=2.4hz,1h),8.04(d,j=7.6hz,2h),7.95(d,j=7.6hz,2h),7.78(d,j=7.6hz,1h),7.24(d,j=7.6hz,2h),6.86(d,j=7.6hz,2h),4.56(t,j=6.4hz,2h),3.84(t,j=6.4hz,2h),3.59(q,j=7.2hz,2h),1.28(t,j=7.2hz,3h).
13
c nmr(cdcl
3
,100mhz)δ165.46,153.04,151.66,147.27,144.55,134.17,134.07,130.68,129.97,126.94,126.47,126.02,122.61,121.83(q,j=273hz),118.04,111.63,62.24,48.84,45.83,28.61,12.37.esi-ms:m/z=561.1[m+h].
[0073]
实施例3:染料(iv)的合成
[0074]
将化合物2替换为化合物4(2mmol,1.118g),其余等同于实施例1,得到染料(iv),蓝色固体,1.05g,收率68.1%。
[0075][0076]
1
h nmr(cdcl
3
,400mhz)δ8.47(dd,j
1
=6.4hz,j
2
=2.0hz,2h),7.74(d,j=7.6hz,2h),7.52(m,4h),6.81(d,j=7.6hz,2h),4.48(t,j=4.4hz,2h),4.06(t,j=4.4hz,2h),3.29(q,j=8.0hz,2h),1.10(t,j=8.0hz,3h).esi-ms:m/z=771.0[m+h].
[0077]
实施例4:染料(v)的合成
[0078][0079]
在氮气保护、0℃条件下,将化合物2(2mmol,0.534g)、氢化钠(60%wt in mineral oil,2mmol,0.080g)和四氢呋喃(20ml)加入三口烧瓶中,搅拌反应30min;将化合物5(2mmol,0.558g)投入上述反应液中,在室温下继续搅拌反应12h;采用薄层色谱法监测反应,12h后原料消耗完毕停止反应。
[0080]
加40ml水、20ml乙酸乙酯,萃取三次(乙酸乙酯40ml
×
3),合并有机相,使用旋转蒸发仪蒸除溶剂,残留物经硅胶柱层析分离(洗脱剂:石油醚/乙酸乙酯=5/1)得到染料(v),黄色固体,0.83g,收率88.7%。
[0081][0082]
1
h nmr(cdcl
3
,400mhz)δ7.82(d,j=7.6hz,2h),7.77(dd,j
1
=7.6hz,j
2
=2.0hz,2h),7.54(dd,j
1
=7.6hz,j
2
=7.2hz,2h),7.47-7.44(m,3h),7.39(d,j=7.6hz,2h),6.69(d,j=7.6hz,2h),4.47(s,2h),3.77(t,j=4.4hz,2h),3.71(t,j=4.4hz,2h),3.29(q,j=8.0hz,2h),1.10(t,j=8.0hz,3h).esi-ms:m/z=468.2[m+h].
[0083]
实施例5:染料(vi)的合成
[0084]
将化合物2替换为化合物6(2mmol,0.629g),其余等同于实施例4,得到染料(vi),红色固体,0.84g,收率82.0%,
[0085][0086]
1
h nmr(cdcl
3
,400mhz)δ8.30(d,j=7.6hz,2h),7.86-7.83(m,4h),7.45(m,4h),6.80(d,j=7.6hz,2h),4.47(s,2h),3.79-3.76(m,2h),3.73-3.69(m,2h),3.29(q,j=8.0hz,2h),1.10(t,j=8.0hz,3h).esi-ms:m/z=512.2[m+h].
[0087]
实施例6:染料(vii)的合成
[0088]
将化合物2替换为化合物7(2mmol,0.842g),其余等同于实施例4,得到染料(vii),蓝色固体,0.93g,收率75.0%。
[0089][0090]
1
h nmr(cdcl
3
,400mhz)δ9.50(s,1h),8.92-8.91(m,2h),8.27(d,j=1.6hz,1h),7.44-7.39(m,5h),6.96(d,j=7.2hz,1h),4.47(s,2h),3.79-3.75(m,2h),3.73-3.69(m,2h),3.29(q,j=8.0hz,2h),2.16(s,3h),1.10(t,j=8.0hz,3h).esi-ms:m/z=620.2[m+h].
[0091]
实施例7:染料(viii)的合成
[0092]
将化合物2替换为化合物8(2mmol,0.394g),其余等同于实施例1,得到染料(viii),黄色固体,0.71g,收率86.7%。
[0093][0094]
1
h nmr(cdcl
3
,400mhz)δ7.89(s,1h),7.76(d,j=7.6hz,2h),7.73-7.70(m,4h),7.55-7.51(m,4h),7.47-7.42(m,1h),7.26(d,j=7.6hz,2h).esi-ms:m/z=410.1[m+h].
[0095]
实施例8:染料(ix)的合成
[0096]
将化合物2替换为化合物9(2mmol,0.642g),其余等同于实施例1,得到染料(ix),红色固体,0.89g,收率83.4%,
[0097][0098][0099]
1
h nmr(cdcl
3
,400mhz)δ8.50(d,j=1.6hz,1h),8.26(dd,j
1
=7.2hz,j
2
=2.0hz,1h),7.89(s,1h),7.87(d,j=7.6hz,1h),7.81(d,j=7.6hz,2h),7.71(d,j=7.2hz,2h),7.54(d,j=7.6hz,2h),7.27(d,j=7.2hz,2h).esi-ms:m/z=533.0[m+h].
[0100]
实施例9:染料(x)的合成
[0101]
将化合物2替换为化合物10(2mmol,0.790g),其余等同于实施例1,得到染料(x),蓝色固体,0.95g,收率78.2%,
[0102][0103]
1
h nmr(cdcl
3
,400mhz)δ8.84(d,j=2.0hz,1h),8.81(d,j=2.0hz,1h),8.70(s,1h),7.73(d,j=7.2hz,2h),7.55(d,j=7.6hz,2h),7.15(d,j=1.6hz,1h),7.07-7.00(m,2h),3.75(s,1h),2.82(s,3h).esi-ms:m/z=607.0[m+h].
[0104]
实施例10:染料(xi)的合成
[0105]
将化合物2替换为化合物8(2mmol,0.394g),其余等同于实施例4,得到染料(xi),黄色固体,0.70g,收率88.5%。
[0106][0107]
1
h nmr(cdcl
3
,400mhz)δ7.73(dd,j
1
=7.6hz,j
2
=2.0hz,2h),7.58-7.51(m,4h),7.47-7.42(m,3h),7.38(d,j=7.6hz,2h),6.75(d,j=7.2hz,2h),4.64(s,1h),4.32(s,
2h).esi-ms:m/z=396.1[m+h].
[0108]
实施例11:染料(xii)的合成
[0109]
将化合物2替换为化合物9(2mmol,0.642g),其余等同于实施例4,得到染料(xii),红色固体,0.87g,收率83.8%。
[0110][0111]
1
h nmr(cdcl
3
,400mhz)δ8.50(d,j=2.4hz,1h),8.26(dd,j
1
=7.6hz,j
2
=2.4hz,1h),7.90(d,j=7.6hz,1h),7.63(d,j=7.6hz,2h),7.45-7.37(m,4h),6.77(d,j=7.6hz,2h),4.56(s,1h),4.32(s,2h).esi-ms:m/z=519.0[m+h].
[0112]
实施例12:染料(xiii)的合成
[0113]
将化合物2替换为化合物11(2mmol,0.806g),其余等同于实施例4,得到染料(xiii),蓝色固体,0.95g,收率78.0%。
[0114][0115]
1
h nmr(cdcl
3
,400mhz)δ9.50(s,1h),8.79(d,j=2.0hz,1h),8.64(d,j=2.0hz,1h),8.78(d,j=7.2hz,1h),7.44(d,j=7.6hz,2h),7.39(d,j=7.6hz,2h),6.71(d,j=2.0hz,1h),6.64(dd,j
1
=7.6hz,j
2
=2.0hz,1h),4.77(s,1h),4.32(s,2h),2.05(s,3h).esi-ms:m/z=601.0[m+h].
[0116][0117]
对比例1:对比染料m的合成
[0118][0119]
以化合物12(2mmol,0.436g)替代实施例1中的化合物1,其余等同于实施例1,得到
对比染料m,黄色固体,0.62g,收率80.8%,
1
h nmr(cdcl
3
,400mhz)δ8.01(d,j=7.6hz,2h),7.73(dd,j
1
=7.6hz,j
2
=2.0hz,2h),7.58-7.51(m,4h),7.47-7.42(m,3h),6.75(d,j=7.6hz,2h),4.65(s,1h),4.32(s,2h).esi-ms:m/z=384.1[m+h].
[0120]
对比例2、对比染料n的合成
[0121]
以化合物13(2mmol,0.776g)替代实施例1中的化合物2,其余等同于实施例1,得到对比染料n。
[0122]
化合物13可参考文献:bioconjugate chemistry,2006,17(3),670-676。
[0123][0124]
染料混合物的实例:染料混合物的制备
[0125]
将染料(ii)2.0g、分散剂nno 2.0g、水20g加入砂磨机中混合后进行高速分散砂磨,砂磨速度为3000转/分,砂磨时间2小时,砂磨结束后将所得分散液进行喷雾干燥(雾化速度为15ml/min,进口温度300℃,出口温度100℃),即得染料混合物,编号1。
[0126]
前述各案例中所得染料以及市售商品染料均按此方法制备染料混合物,采用染料(iii)至染料(xiii)所得染料混合物的编号分别为2~12,采用对比染料m所得染料混合物对应编号13,采用对比染料n所得染料混合物对应编号14,采用市售商品染料c.i.分散黄23所得染料混合物对应编号15;采用市售商品染料活性分散黄gr所得染料混合物对应编号16。
[0127]
c.i.分散黄23的结构为:
[0128][0129]
活性分散黄gr的结构为:
[0130][0131]
实验一:取编号1的染料混合物0.040g溶于去离子水100ml,配制成水溶液,放置于染缸中,采用醋酸将水溶液的ph调节至5,加入作为促染剂的丁二酸二丁酯0.1g;放入涤纶织物2g,密封染缸,以2℃/min的速率升温至95℃,保温30min;染色结束后,将染缸自然冷却至室温,取出织物,用清水冲洗,晾干后,放入160℃的焙烘箱中焙烘2h,得染色涤纶织物;采用data color测色配色仪测试染色织物表面色深值(k/s)。
[0132]
说明:本发明加入促染剂的目的是使染料在较低温度下也能很好地进入涤纶纤维内部;进一步的目的,是为了降低染色时温度,保证在复杂的染色环境里染料结构中的双吖丙啶能够稳定存在;染色后再把助剂从纤维上洗掉,晾干(去除水分),因此在焙烘时仅有染料和涤纶纤维,以此保证固色率。
[0133]
实验二:将实验一所得的染色涤纶织物放入装有n,n-二甲基甲酰胺(100ml)的单口烧瓶中,加热至120℃,保持30min,取出织物冷却,用清水冲洗,晾干,测试织物k/s值。编号2~16的染料混合物均按实验一和实验二方法进行实验,结果见表1:
[0134]
表1
[0135][0136][0137]
其中,固色率计算方法为:固色率=(实验二溶剂萃取后染色织物k/s值)/(实验一染色织物k/s值)。
[0138]
由表1结果可以看出,染料(ii)~染料(xiii)所染涤纶织物具有较好的得色率,经n,n-二甲基甲酰胺萃取后仍然有大部分染料残留于纤维上,固色率为60%以上,同时也可以看出,随着染料分子结构中n-h键数量的增加,固色率由约80%下降到不及65%。编号13结果表明,染料分子中去除双吖丙啶结构后,即无反应能力,染色织物上的染料被n,n-二甲基甲酰胺全部萃取下来;编号14结果表明,染料分子结构中含有多个羟基时,固色率同样会有显著降低,猜测原因可能是卡宾中间体与染料自身结构上的羟基发生了反应而未能与纤维结合;编号15结果表明,商品分散染料c.i.分散黄23与涤纶纤维间亦无共价键,纤维上染
料能够被n,n-二甲基甲酰胺全部萃取下来;编号16结果表明,含氯代三嗪活性基的市售活性分散染料与涤纶纤维间亦不能形成共价键;上述结果间接证明了本发明提供的染料能够与涤纶纤维有效发生共价键结合。
[0139]
实验三:按照标准gb/t 3921-2008、gb/t 3920-2008和gb/t 6152-1997测试实验一中各染色织物的耐皂洗色牢度、耐摩擦色牢度和耐升华色牢度。结果见表2。
[0140]
表2
[0141][0142][0143]
由表2结果可知,本发明提供的反应性分散染料染色织物具有很好的耐皂洗、耐摩擦及耐升华色牢度。
[0144]
对比实验1、采用编号1染料混合物进行实验一和实验二,将实验一中保温温度改为70℃,其余等同。
[0145]
对比实验2、采用编号1染料混合物进行实验一和实验二,将实验一中保温温度改为120℃,其余等同。
[0146]
对比实验3、采用编号1染料混合物进行实验一和实验二,将实验一中保温时间改为10min,其余等同。
[0147]
对比实验4、采用编号1染料混合物进行实验一和实验二,将实验一中保温时间改
为60min,其余等同。
[0148]
对比实验5、采用编号1染料混合物进行实验一和实验二,将实验一中焙烘温度改为120℃,其余等同。
[0149]
对比实验6、采用编号1染料混合物进行实验一和实验二,将实验一中焙烘温度改为140℃,其余等同。
[0150]
对比实验7、采用编号1染料混合物进行实验一和实验二,将实验一中焙烘温度改为180℃,其余等同。
[0151]
对比实验8、采用编号1染料混合物进行实验一和实验二,将实验一中焙烘时间改为0.5h,其余等同。
[0152]
对比实验9、采用编号1染料混合物进行实验一和实验二,其中,实验一在染色结束后不进行焙烘,其余等同。
[0153]
对比实验10、采用编号1染料混合物、并采用当前最为流行的针对涤纶的常规高温高压染色法进行染色。取编号1的染料混合物0.040g溶于去离子水100ml,配制成水溶液,放置于染缸中,采用醋酸将水溶液的ph调节至5;放入涤纶织物2g,密封染缸,以2℃/min的速率升温至130℃,保温60min;染色结束后,将染缸自然冷却,取出织物,用清水冲洗,得染色涤纶织物;采用data color测色配色仪测试染色织物表面色深值(k/s)。并进行实验二。
[0154]
对比实验11、将对比实验10中采用高温高压染色法所得染色涤纶织物(进行实验二前的织物)先在160℃的焙烘箱中焙烘2h;再采用data color测色配色仪测试染色织物表面色深值(k/s)。并进行实验二。
[0155]
将对比实验1至对比实验11中的染色织物及萃取后染色织物测试k/s值,结果见表3:
[0156]
表3
[0157][0158]
由表3结果可知,对比实验1表明,染色时保温温度不足时,染料较少上染到涤纶纤维上,导致染色不深;对比实验2表明,染色时温度过高,固色率反而下降,猜测原因可能是由于温度过高导致生成卡宾中间体与水反应而失活;对比实验3表明,保温时间较短时,染料染得涤纶纤维色深值不高;对比实验4表明,保温时间过长,固色率有所下降;对比实验5和6表明,焙烘温度不足时,固色率较低;对比实验8表明,焙烘时间不足时,固色率较低;对比实验9表明,不进行焙烘处理时,染料无法与纤维以共价键结合;对比实验10表明,采用分散染料常规高温高压法进行染色也很难将双吖丙啶染料与涤纶纤维结合,猜测原因可能是高温导致生产卡宾中间体与水反应而失活;对比实验11表明,经高温高压染色法所染涤纶织物即使再进行焙烘处理也无法使染料与纤维再结合,进一步表明双吖丙啶染料已失活。
[0159]
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签: 染料

 热门咨询
热门咨询




tips