
 商标分类
商标分类  商标转让
商标转让 
一种磷酸根响应的碳量子点的制备及其用于指纹荧光识别的应用的制作方法
 2021-02-02 14:02:32|
2021-02-02 14:02:32| 327|
327| 起点商标网
起点商标网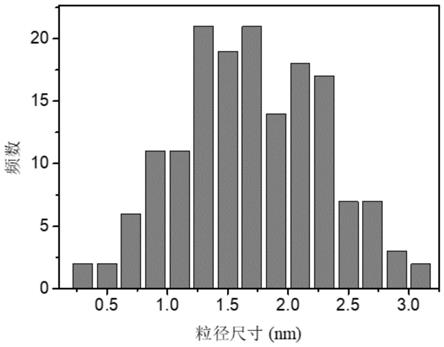
[0001]
本发明涉及碳量子点应用技术领域,特别涉及一种磷酸根响应的碳量子点的制备及其用于指纹荧光识别的应用。
背景技术:
[0002]
指纹作为重要的个人信息,已广泛应用于法医学、刑侦、生物识别和医疗诊断等领域,因为指纹对于每个人都是唯一的,且不会随年龄增长而变化(analytical methodse,2016,8:6293-6297)。在许多情况下,指纹是不可见的(潜指纹),只有在用特定方法处理后才能肉眼直接看到它们(journal of colloid and interface science,2018,518:200-215)。因此,开发指纹“可视”的新技术对于不可见指纹的可视化辨别至关重要。通常,手指表面汗孔分泌出的汗液包含多种成分,如磷酸盐(pi),氯化物,脂质,氨基酸等,它们通过表皮孔到达手指表面,这些分泌物在指纹的可视化或显影成像中起着重要作用(analytical chemistry,2019,91:11185-11191)。目前,已有的用于可视化成像指纹的方法有荧光、拉曼、傅立叶变换红外、光声成像等。其中,荧光的方法是一种灵敏度高、选择性好、检出限低的微量分析方法,它克服了传统检测手段步骤繁琐、耗费时间长等缺点,可以实时对待测物进行定性和定量检测。
[0003]
碳量子点(cqd)由于具有独特的理化特性,如强的光致发光、良好的生物相容性、结构易修饰等,因此在生物成像、传感、药物输送、催化、能量转换和癌症治疗等领域中被广泛应用。目前的cqd用于指纹成像,主要依靠cqd与手指汗孔分泌组分之间的静电作用。这种相互作用比较弱,不利于潜指纹的精细结构成像。另外cqd与手指汗孔分泌组分之间也可以借助金属离子如cu
2+
,fe
3+
,hg
2+
,fe
2+
或pb
2+
进行相互作用成像指纹(analytical and bioanalytical chemistry,2020,412:1317-1324)。这种成像方法需要借助有毒金属离子,同时需要多重参数之间的优化,不利于大规模生成和应用。开发选择性识别手指汗孔分泌的主要组分-磷酸(pi)的cqd,并基于此作用进一步开发用于潜指纹荧光成像的cqd,构建成像潜指纹的新方法,这在磷酸的检测、法医、刑侦上具有重要意义。
技术实现要素:
[0004]
为了克服以上技术问题,本发明的目的在于提供一种磷酸根响应的碳量子点的制备及其用于指纹荧光识别的应用,通过水热法制备磷酸根响应的碳量子点,具有快速、简便、高效,毒性低的特点,该碳量子点作为增强型荧光探针用于磷酸根的定性和在一定浓度范围内的定量检测,同时借助碳量子点与磷酸的直接作用来增强荧光这一机制,该碳量子点应用于纸上潜指纹的三级(level 3)荧光成像。
[0005]
为了实现上述目的,本发明采用的技术方案是:
[0006]
一种磷酸根响应的碳量子点的制备方法,包括以下步骤;
[0007]
步骤1:将0.074~0.3mmol 1,2,4-苯三酚加入50ml超纯水中,搅拌3分钟,得到1,
2,4-苯三酚溶液;
[0008]
步骤2:将步骤1所得的溶液装入内衬聚四氟乙烯的高压反应釜中,在150~180℃下反应4~8小时;反应完成后在8000~12000r/min转速下离心,取上层清液待用;
[0009]
步骤3:将步骤2所得的上层清液进行旋转蒸发、真空冷冻干燥,得到黑色固体粉末,该黑色固体粉末为所述碳量子点。
[0010]
所述1,2,4-苯三酚的结构为:
[0011]
一种磷酸根响应的碳量子点应用于指纹荧光识别方法,包括以下步骤;
[0012]
步骤1:将得到的碳量子点粉末0.3mg加入到1ml去离子水中,配置成0.3mg/ml母液待用;
[0013]
步骤2:取步骤1得到的母液200μl加入2.8ml超纯水中制备得到20μg/ml的碳量子点溶液;
[0014]
步骤3:将步骤2得到的量子点溶液喷洒在15cm
×
15cm的滤纸上,将该滤纸在45℃~60℃温度下烘干备用;
[0015]
步骤4:将手指按在步骤3所得的滤纸上,5秒后在可见光下观察该滤纸,滤纸上无指纹成像,在365nm紫外灯下明显地观察到蓝色的荧光指纹图案。
[0016]
所述步骤1中得到的碳量子点母液发出微弱的蓝色荧光,其最大激发波长为370nm,最大发射峰位于464nm处,在场发射透射电镜下观察,该碳量子点大小为0.31~3.19nm,平均粒径约1.7
±
0.38nm,在高分辨透射电镜观察,该碳量子点的晶面间距为0.21nm,对应石墨碳的100晶面,在xrd图谱中制得的碳量子点具有26
°
为中心的特征峰,该峰属于石墨化碳的(002)面。
[0017]
所述步骤2中得到的碳量子点溶液中分别加入各种离子、氨基酸进行荧光测试,只有加入po
43-后,该碳量子点才能发出强烈的蓝色荧光,其最大激发波长为370nm,最大发射峰位于448nm处。
[0018]
所述步骤2中得到碳量子点溶液应用于磷酸根的定性和在一定浓度范围内(0~100μmol/l)的定量检测。
[0019]
所述步骤2得到的量子点溶液用于指纹的荧光成像,普通滤纸经过碳量子点喷洒干燥后,借助碳量子点与磷酸的直接作用来增强荧光这一机制,手指按压该滤纸5s后,在365nm紫外光下可以观察到明显的蓝色荧光指纹图案,该指纹图案包括清晰的level 1、level 2和level 3在内的指纹细节。
[0020]
本发明的有益效果:
[0021]
本发明以1,2,4苯三酚为碳源,采用水热法合成了碳量子点。该碳量子点的制备工艺简单、成本低廉、稳定性好,在超纯水中没有荧光。
[0022]
加入po
43-后,能够立刻发出明显的蓝色荧光。该碳量子点可以作为增强型的荧光探针选择性地检测po
43-。该碳量子点对其它常见的离子、氨基酸没有明显的荧光变化响应,可用于纯水中po
43-的定性和在一定浓度范围内(0~100μmol/l)定量检测。该碳量子点对
po
43-的检测具有很好的灵敏性。
[0023]
本发明合成得到的碳量子点在纸上自身荧光较弱,只有与po
43-作用后有显著的蓝色荧光增强。该碳量子点溶液喷洒过的纸可用于指纹的三级(level 3)荧光成像,为纸质文件安全提供可能。
附图说明
[0024]
图1是制备的碳量子点的粒径分布图。
[0025]
图2是制备的碳量子点的透射电镜图,内嵌图为放大的晶面图。
[0026]
图3是制备的碳量子点的x射线衍射图。
[0027]
图4是制备的碳量子点的母液的荧光激发和发射光谱图。
[0028]
图5是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在浓度260μmol/l po
43-存在前后的荧光激发光谱和发射光谱图。
[0029]
图6是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在分别加入浓度0-180μmol/l po
43-后在448nm处的荧光强度的变化图。
[0030]
图7是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在加入浓度0-180μmol/l po
43-后在448nm处的对数荧光强度值(以10为底数)随不同po
43-浓度的线性关系图。
[0031]
图8是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在分别加入浓度140μmol/l常见阳离子后448nm处的荧光强度图。
[0032]
图9是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在分别加入浓度140μmol/l的常见阳离子后在448nm处的荧光强度柱状图。
[0033]
图10是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在分别加入浓度140μmol/l常见阴离子后在448nm处的荧光强度图。
[0034]
图11是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在分别加入浓度266.67μmol/l常见阴离子后在448nm处的荧光强度柱状图。
[0035]
图12是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在分别加入浓度140μmol/l常见生物分子后在448nm处的荧光强度图。
[0036]
图13是在ph为7.4的去离子水中,浓度为20μg/ml碳量子点在分别加入浓度140μmol/l常见生物分子后在448nm处的荧光强度柱状图。
[0037]
图14是喷有碳量子点的滤纸被手指按压5秒后在365nm紫外光下的荧光指纹图案以及包括level 1、level 2和level 3在内的指纹细节图,标尺为1cm。
具体实施方式
[0038]
下面结合实施例对本发明作进一步详细说明。
[0039]
磷酸根响应的碳量子点的制备。
[0040]
实施例1:
[0041]
1.在100ml的烧杯中加入50ml超纯水,然后加入0.15mmol的1,2,4-苯三酚,磁力搅拌3分钟,得到1,2,4-苯三酚溶液。
[0042]
2.将步骤1所得的1,2,4-苯三酚溶液装入内衬聚四氟乙烯的高压反应釜中,在170℃下反应6小时,反应完成后在8000r/min下离心,取上层清液待用。
[0043]
3.将步骤2所得上层清液进行旋转蒸发、真空冷冻干燥,得到黑色固体粉末。
[0044]
4.将步骤3所得固体粉末0.3mg溶解在1ml超纯水中,获得母液待用。
[0045]
5.将母液进行在场发射透射电镜下观察,该碳量子点大小为0.31~3.19nm,平均粒径约1.7
±
0.38nm(见附图1)。获得的固体粉末进行高分辨透射电子显微镜测试,粒子成近球形,碳量子点的晶面间距分别为0.21nm(见附图2),分别属于石墨化碳的100晶面。
[0046]
6.将获得的固体粉末进行xrd测试,合成的粉末具有以26
°
为中心的特征峰(见附图3),该峰属于石墨化碳的(002)面。所述的固体粉末为碳量子点粉末。
[0047]
7.将步骤4所得碳量子点母液进行荧光性能测试,其具有较弱的荧光,最大激发波长为370nm,最大发射波长为464nm(见附图4)。
[0048]
实施例2:
[0049]
1.在100ml的烧杯中加入50ml超纯水,然后加入0.074mmol的1,2,4-苯三酚,磁力搅拌3分钟,得到1,2,4-苯三酚溶液。
[0050]
2.将步骤1所得的1,2,4-苯三酚溶液装入内衬聚四氟乙烯的高压反应釜中,在150℃下反应4小时,反应完成后在8000r/min下离心,取上层清液待用。
[0051]
3.将步骤2所得上层清液进行旋转蒸发、真空冷冻干燥,得到黑色固体粉末。
[0052]
4.将步骤3所得固体粉末0.3mg溶解在1ml超纯水中,获得母液待用。
[0053]
实施例3:
[0054]
1.在100ml的烧杯中加入50ml超纯水,然后加入0.3mmol的1,2,4-苯三酚,磁力搅拌3分钟,得到1,2,4-苯三酚溶液。
[0055]
2.将步骤1所得的1,2,4-苯三酚溶液装入内衬聚四氟乙烯的高压反应釜中,在180℃下反应8小时,反应完成后在12000r/min下离心,取上层清液待用。
[0056]
3.将步骤2所得上层清液进行旋转蒸发、真空冷冻干燥,得到黑色固体粉末。
[0057]
4.将步骤3所得固体粉末0.3mg溶解在1ml超纯水中,获得母液待用。
[0058]
制备的碳量子点对磷酸根定性和定量检测的应用。
[0059]
1.取200μl 0.3mg/ml的母液加入2.8ml超纯水中,再加入260μmol/l的po
43-进行荧光性能测试,其最大激发波长为370nm,最大发射波长为448nm(见附图5)。
[0060]
2.将0.04mg碳量子点固体粉末加入3ml超纯水(ph=7.4)中,向其溶液中分别加入浓度为0,10,20,30,40,50,60,70,80,90,100,110,120,130,149,150,160,170,180μmol/l的po
43-,检测其荧光光谱(见附图6)。在加入20~180μmol/l po
43-后其在发射波长448nm处的荧光强度的对数值(以10为底数)随不同po
43-浓度具有良好的线性关系,拟合方程为y=0.01015x-3.1757,相关系数r2=0.996,检出限为0.34μmol/l(见附图7)。
[0061]
3.将0.04mg碳量子点固体粉末加入3ml超纯水(ph=7.4)中,向其溶液中分别加入浓度为140μmol/l的ag
+
,ba
2+
,ca
2+
,cd
2+
,cr
3+
,cu
2+
,fe
2+
,fe
3+
,hg
2+
,k
+
,mg
2+
,mn
2+
,ni
2+
和zn
2+
,检测荧光强度变化(见附图8)。只有加入po
43-后才能在448nm处检测到蓝色荧光,而加入以上这些常见的阳离子物质没有响应。加入不同阳离子分别观察在448nm处的相对荧光强度变化(图9)。
[0062]
4.将0.04mg碳量子点固体粉末加入3ml超纯水(ph=7.4)中,向其溶液中分别加入浓度为140μmol/l的f-,cl-,br-,co
32-,hco
3-,i-,no
3-,so
32-和po
43-,检测荧光强度变化(见附图10)。只有加入po
43-后才能在448nm处检测到蓝色荧光,而加入以上这些常见的阴离子物
质没有响应。加入不同阴离子后分别观察在448nm处的相对荧光强度变化(图11)。
[0063]
5.将0.04mg碳量子点固体粉末加入3ml超纯水(ph=7.4)中,向其溶液中分别加入浓度为140μmol/l的l-苯丙氨酸,l-丙氨酸,dl-蛋氨酸,甘氨酸,亮氨酸,色氨酸,l-苏氨酸,缬氨酸,组氨酸,高半胱氨酸,葡萄糖和半胱氨酸,检测荧光强度变化(见附图12)。只有加入po
43-后才能在448nm处检测到蓝色荧光,而加入以上这些常见的氨基酸物质没有响应。加入不同氨基酸后分别观察在448nm处的相对荧光强度变化(图13)。
[0064]
制备的碳量子点用于纸上指纹荧光识别的应用。
[0065]
1.将0.08mg碳量子点固体粉末加入3ml超纯水(ph=7.4)中,获得26.6μg/ml的碳量子点溶液。
[0066]
2.将获得的溶液喷在15cm
×
15cm的滤纸上,将该滤纸置于45℃烘箱中烘干。用手指按压在该滤纸上5秒。自然光下,观察不到滤纸上的指纹。在365nm紫外光下可以明显地观察到蓝色荧光指纹图案。该指纹图案包括清晰的指纹轮廓-螺纹(level 1)、短脊,脊分叉(level 2)和汗孔(level 3)在内的指纹细节(见附图14)。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips