
 商标分类
商标分类  商标转让
商标转让 
稠合吡啶杂环的无金属无溶剂合成及其生物医学应用的制作方法
 2021-02-02 14:02:06|
2021-02-02 14:02:06| 516|
516| 起点商标网
起点商标网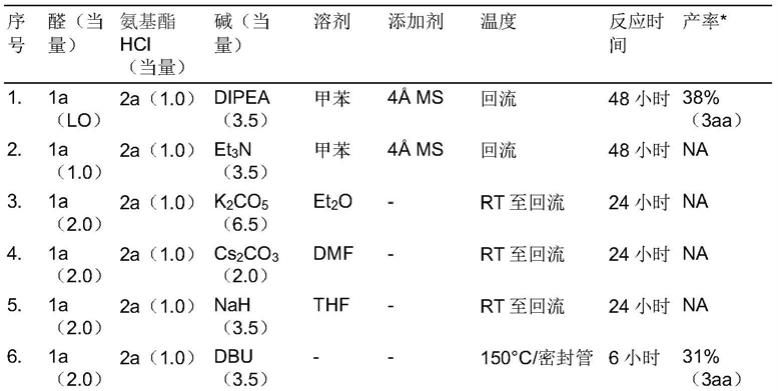
稠合吡啶杂环的无金属无溶剂合成及其生物医学应用
发明领域
[0001]
本发明涉及一种稠合吡啶杂环的无金属无溶剂合成方法和系统及其在癌症和结核病治疗中的应用。
背景技术:
[0002]
稠合吡啶类化合物属于最大的生物碱家族,并且广泛分布于自然界中,包括植物、海洋生物、昆虫、哺乳动物以及人体组织和体液中。这些杂环化合物因其多样的生物活性而备受关注。它们充当特定蛋白质的特异性配体,且借助这些外部配体(激动剂和拮抗剂)谨慎地调节蛋白质的表达,可以操纵重要的生物学功能。因此,为每一个新发现的分子寻找合适的蛋白质-配体相互作用在化学生物学领域中是一个重要课题。将新合成的分子与已存在的(已知的)配体进行结构类比,可能成为解开这个谜团的重要工具。
[0003]
氮吲哚(吡咯吡啶)是吲哚和嘌呤的最接近生物等排体。因此,它们在模拟天然配体-靶相互作用中起着不可或缺的作用。因此,吡咯吡啶已成为近年来开发的多种重要候选药物中不可或缺的核心单元。然而,它们的合成一直是合成化学行业的一大挑战。由于吡啶环的缺电子性质,经典的吲哚合成(包括fischer环化和madelung环化)往往不能有效地应用于相应氮吲哚的合成。另外,制药业普遍缺乏常规开发的方案,因为合成策略中的任何重金属都可能导致有害的毒性和生物学研究的不准确性。
[0004]
吲哚吡啶(咔啉)广泛具有dna插层特性、酶抑制特性(主要是cdk、拓扑异构酶和单胺氧化酶)以及与苯二氮卓受体和5-羟基5-羟色胺受体的相互作用。此外,这些化合物已显示出光谱的药理学性质,包括镇静、抗焦虑、催眠、抗惊厥、抗肿瘤、抗病毒、抗寄生虫以及抗菌活性。尽管如此,但定制咔啉衍生物的合成对合成化学行业仍然是尚未解决的问题。对现有方案的总体评估揭示了以下严重问题:产率低、底物范围有限(包括使用非常特殊的起始材料)、涉及极端热条件、腐蚀性试剂和有毒重金属催化剂。
[0005]
在过去的二十年里,呋喃并吡啶类化合物作为吲哚的生物等排体得到了广泛的研究。因此,这些杂环化合物在认知或自身免疫性疾病、偏头痛、肠易激综合征和哮喘等治疗领域成为有用的药效基团。苯并呋喃并吡啶,作为稠合吡啶类的另一引人注目的成员,在制药和oled工业中也引起了引人注目的应用。然而,由于底物范围有限且缺乏创新方法,合成方法的进一步发展仍然受到限制。现有的方法主要依赖于昂贵的重金属催化剂,并且仅限于取代的吡啶衍生物上的呋喃环合成。在呋喃衍生物上形成吡啶核的替代方法尚未研究透彻。
[0006]
对现有技术方法的挑战进行总结,由于固有的结构复杂性,标准的吲哚和咔唑合成方案不适用于该领域。相比于苯对应物,吡啶环非常缺失电子。这限制了吡啶环上形成五元环的范围。此外,由于底物范围较窄,尚未彻底研究吡咯和吲哚上含氮六元环的形成。同样,吡咯和吲哚衍生物的热稳定性也限制了它们作为起始材料的用途。此外,基质范围窄、起始材料昂贵、重金属催化剂和工作程序繁琐的方法很难商业化,因为这些元素在大规模生产中不具有成本效益。此外,常规方法采用金属催化剂,有机溶剂被广泛应用于有机合成
中,并且由于其先天毒性和环境危害而备受关注。
[0007]
为了克服上述缺点,已在本文中设计并开发了一种多用途的一锅法来合成一系列取代氮杂环。新颖的合成方案是一种无金属且无溶剂的方法,只需对起始材料进行策略设计就可以得到多种化合物。对合成的化合物进行仔细筛选,发现其具有抗癌、抗分枝杆菌和抗生物膜化合物等有意义的生物活性或功效。通过本发明合成的化合物不需要复杂的纯化程序或昂贵的精密设备。发明目的
[0008]
本文实施例的主要目的是提供稠合吡啶杂环。
[0009]
本发明的另一个目的是提供一种无金属无溶剂的稠合吡啶杂环合成方法。
[0010]
本发明实施例的另一个目的是合成具有抗癌症和抗多药耐药病原体生物功效的化合物。
技术实现要素:
[0011]
因此,本文中的实施例提供稠合吡啶杂环及其制备方法。本发明所述吡啶杂环可用于治疗癌症(宫颈、肾脏、肺、乳房和表皮皮肤)和耐多药结核病。这些杂环还能够用作抗生物膜剂限制致病株,从而将继发感染的风险降至最低。
[0012]
在一个实施例中,所述稠合吡啶杂环,例如式i氮吲哚、咔啉衍生物、呋喃并[b]吡啶或呋喃并[b]吡啶-靛红杂合体。及其前药、立体异构体、外消旋体、盐、水合物、水合盐、酸盐水合物、溶剂化物、同构晶形及其组合物;其中,
‘
x
’
是氮或氧中的一种;r
1
是吗啉甲酮、-conh
2
、-cn、-cho、-cooh、-roh、-coor中的一种,其中r是烷基r
2
和r
5
独立地选自由-h、-boc、烷基、对甲苯磺酰基、苯基磺酰基、芳氧基、苄氧基、任选取代苄基和任选取代芳基组成的组中的至少一种;r
3
、r
4
、r
6
和r
7
独立地选自-h、烷基、或r
3
和r
4
中的至少一种;并且r
6
和r
7
能够稠合以形成任选取代的苯环。
[0013]
在另一个实施例中,本发明还提供了一种用于制备嘧啶类稠合杂环的方法,所述方法包括:使选自由n-取代吡咯-2-甲醛、n-取代吲哚-甲醛、任选取代的呋喃甲醛以及苯并呋喃-2-吡咯甲醛组成的组的反应物与甘氨酸烷基酯的酸性盐在碱性、100至160℃温度范
围条件下接触3至15小时,获得式i化合物。
[0014]
当结合以下描述和附图考虑时,将更好地领会和理解本文所述实施例的这些和其他方面。然而,应当理解的是,在指示优选实施例及其众多具体细节的同时,以下描述是通过说明而不是限制的方式给出的。在不脱离其主旨的前提下,可以在本文所述实施例的范围内进行多种变更和修改,并且本文所述实施例包括所有类似变更。
附图说明
[0015]
该方法和系统在附图中进行了说明,在附图中,类似的参考字母指示了各个图中的相应部分。通过以下参考附图的描述,可以更好地理解本文所述实施例,其中:
[0016]
图1展示了根据本公开所述实施例的发明的示意图;
[0017]
图2展示了根据本公开所述实施例,从n-取代吡咯-2-醛1a-i合成5-氮杂吲哚3aa-fa;
[0018]
图3展示了根据本公开所述实施例,1-取代-2-吡咯醛(1a-i)的1,4,5-位上的各种取代基在形成5-氮吲哚3aa-fa中的作用;
[0019]
图4展示了根据本公开所述实施例,5-氮吲哚(3aa-fa)、咔啉(18aa-fa)和氮杂苯并呋喃(20aa-ba)和吡咯苯并呋喃(28aa-ac)的合理形成机制;
[0020]
图5展示了根据本公开所述实施例,对各种杂环的5-区域单体而不是6-区域异构体的形成的说明;
[0021]
图6展示了根据本公开所述实施例,5-氮吲哚衍生物3aa(ccdc 1836867)的单晶xrd分析;
[0022]
图7展示了根据本公开所述实施例,新型5-氮吲哚衍生物(11至14)的合成;
[0023]
图8展示了根据本公开所述实施例,拟定cb2激动剂(16)的合成;
[0024]
图9展示了根据本公开所述实施例,γ-咔啉18aa-fa的合成;
[0025]
图10展示了根据本公开所述实施例,γ-咔啉衍生物18ac(ccdc:1897787)的单晶xrd分析;
[0026]
图11展示了根据本公开所述实施例,呋喃并[b]吡啶(20aa-ba)的合成;
[0027]
图12展示了根据本公开所述实施例,用于抗结核活性的呋喃吡啶-靛红杂合物(26)的合成;
[0028]
图13展示了根据本公开所述实施例,苯并呋喃并吡啶(28aa-ac)的合成;
[0029]
图14展示了根据本公开所述实施例,苯并呋喃并吡啶(28aa-ac)的合成;
[0030]
图15展示了根据本公开所述实施例,18ac在dmso中(左侧;λexc 360nm)和10-5m化合物18ac溶液在四种不同溶剂中在紫外室内(右侧)的荧光衰减曲线;
[0031]
图16展示了咔啉18ac、18bc、18da和18fa在人癌细胞系hela、mcf-7、hek293、a431和a549中的剂量-反应曲线。根据本公开所述实施例,通过一式三份的标准结晶紫分析测定ic50值;
[0032]
图17展示了根据本公开所述实施例,用于hela细胞中18ac摄取的共焦显微镜研究(λex=405nm;收集范围=420至470nm);
[0033]
图18展示了根据本公开所述实施例,各种化合物的结构式;
[0034]
图19展示了根据本公开所述实施例,将本发明与现有方法区分开的本发明的关键
特征。
[0035]
图20展示了根据本公开所述实施例,呋喃并[b]吡啶-靛红杂合物衍生物26针对耻垢分枝杆菌(50至100μm)的体外抗结核活性,sd(n=3);耐多药/机会致病性牛分枝杆菌(70至100μm)sd(n=3)。
具体实施方式
[0036]
参考附图中示出并在以下描述中详细说明的非限制性实施例,更全面地解释本文中的实施例及其各种特征和有利细节。省略对公认组分和处理技术的说明,以便清晰本文中的实施例。另外,本文描述的各种实施例不一定相互排斥,因为某些实施例可以与一个或多个其他实施例组合形成新的实施例。除非另有说明,本文使用的术语“或”是指非排他性或。本文使用的示例仅仅是为了便于理解本文所述实施例可以被实践的方式,并且进一步使本领域技术人员能够实践本文所述实施例。因此,不应将示例解释为限制本文所述实施例的范围。
[0037]
如本文所用术语“烷基”包括碳原子链,该链是任选支链。
[0038]
本文所用术语“芳基”包括分子片段或自由基,其包含碳原子的芳香单环或多环环,例如苯基、萘基诸如此类。
[0039]
本文所用术语“取代芳基”包括分子片段或自由基,其包含具有一个或多个取代基的芳基,例如烷基、杂烷基、卤素、羟基、氨基、烷基或二烷基氨基、烷氧基、烷基磺酰基、氨基磺酰基、羧酸盐、烷氧羰基、氨基羰基、氰基、硝基诸如此类。应理解,可任选地用卤素取代此类取代基中的所述烷基。
[0040]
本文所述发明涉及吡啶稠杂环的发现,例如取代的5-氮吲哚、γ-咔啉、呋喃并[b]吡啶、5-氮吲哚或呋喃并[b]吡啶-靛红杂合物、苯并呋喃并吡啶及其硫系对应物。本发明的化合物在治疗癌症[hela(宫颈癌)、mcf-7(乳腺癌)、hek293(肾)、a431(宫颈鳞状上皮)和a549细胞(肺)]和耐多药病原体,例如引起结核病的剂(耻垢分枝杆菌、牛分枝杆菌(bcg)和其他致结核株方面具有应用价值。本发明的化合物还可用于针对生物膜形成菌或病原体组的抗生物膜活性。本发明还提供了一种合成吡啶稠杂环的简便方法。该方法是一种简单的一锅一步法获得多种杂环的通用方法。产品纯化简单,产率具有可重复性。
[0041]
本发明所述方法描述了一种简单的一锅法合成吡啶呋喃衍生物库。此外,在我们实验室还开发了一组新型吡啶呋喃与靛红结合的靛红杂合物,并证明其对引起结核病的非致病性和致病性分枝杆菌都具有抗分枝杆菌活性。此外,还研究了几种新型吡啶呋喃分子及其功能衍生物的抗tb性能,并证明其对耐多药分枝杆菌株具有抗结核活性。
[0042]
所发明的合成方法已经成功优化,能够通过简化的一锅法以可观的产率生产各种杂环,否则只能通过多步合成以非常有限的产率制备这些杂环。
[0043]
在本发明的一个实施例中,所述稠合吡啶杂环,例如氮吲哚、咔啉衍生物、呋喃并[b]吡啶是式i化合物
及其前药、立体异构体、外消旋体、盐、水合物、水合盐、酸盐水合物、溶剂化物、同构晶形及其组合物;其中,
‘
x
’
是氮或氧中的一种;r
1
是吗啉甲酮、-conh
2
、-cn、-cho、-cooh、-roh、-coor中的一种,其中r是烷基r
2
和r
5
独立地选自由-h、-boc、烷基、对甲苯磺酰基、苯基磺酰基、芳氧基、苄氧基、任选取代苄基和任选取代芳基组成的组中的至少一种;r
3
、r
4
、r
6
和r
7
独立地选自-h、烷基、或r
3
和r
4
中的至少一种;并且r
6
和r
7
能够稠合以形成任选取代的苯环。
[0044]
在另一个实施例中,本发明公开了式i化合物,其中x是氮;r
1
是吗啉甲酮、-conh
2
、-cn、-cho、-cooh、、-roh和-coor中的一种,其中r是选自由-me、et或
t
bu组成的组的烷基;r
2
与r
5
相同,并且选自由氢、甲基、苄基、甲氧基苄基、对甲苯磺酰基和苯磺酰基组成的组;r
3
、r
4
、r
6
和r
7
是独立地选自-h或-ch
3
的一种。
[0045]
在另一个实施例中,本发明公开了式i化合物,其中x是氮;r
1
是吗啉甲酮、-conh
2
、-cn、-cho、-cooh、、-roh和-coor中的一种,其中r是选自由-me、et或
t
bu组成的组的烷基;r
2
与r
5
相同,并且选自由氢、甲基、苄基、甲氧基苄基、对甲苯磺酰基和苯磺酰基、r
3
和r
4
组成的组;且r6和r7能够稠合以形成任选取代的苯环。
[0046]
在另一个实施例中,所述式i化合物包括其中r
1
是-coor,其中r是选自由-me、et或
t
bu组成的组的烷基;r
3
、r
4
、r
6
和r
7
是独立地选自-h、烷基或r
3
和r
4
中的至少一种;且r
6
和r
7
能够稠合以形成任选取代的苯环。
[0047]
在另一个实施例中,本发明公开了式ii化合物
及其前药、立体异构体、外消旋体、盐、水合物、水合盐、酸盐水合物、溶剂化物、同构晶形及其组合物;其中r1是任选取代的靛红;r3、r4、r6和r7是独立地选自-h、烷基或r3和r4中的至少一种;且r6和r7能够稠合以形成任选取代的苯环。
[0048]
在一个实施例中,所述式ii化合物是
[0049]
本发明还提供了一种制备所述式i和ii化合物的简便方法。在一个实施例中,所述式i化合物可以通过以下步骤制备:将选自由n-取代吡咯-2-甲醛、n-取代吲哚-甲醛、任选取代呋喃甲醛和苯并呋喃-2-甲醛组成的组的反应物与甘氨酸烷基酯的酸性盐在碱(选自n,n-二异丙基乙胺(dipea)、三乙胺(et
3
n)、k
2
co
3
、nah、cs
2
co
3
和1,8-二氮杂二环[5.4.0]十一碳-7-烯(dbu))存在条件下、100至160℃温度范围条件下接触3至15小时,获得所述式i化合物在所述实施例中,反应物与甘氨酸烷基酯酸性盐的摩尔比介于1:2至2:1的范围内,优选地是2:1,并且反应物与碱的摩尔比介于1:1至1:15的范围内,优选地介于1:1至1:2的范围内。
[0050]
本发明还公开了一种制备式ii化合物的方法。该方法包括以下步骤:将任选取代的furfural与甘氨酸烷基酯的酸性盐在碱(选自n,n-二异丙基乙胺(dipea)、三乙胺(et
3
n)、k
2
co
3
、nah、cs
2
co
3
和1,8-二氮杂二环[5.4.0]十一碳-7-烯(dbu))存在条件下、100至160℃温度范围条件下接触3至15小时,获得所述式i化合物;b)在第一反应条件(温度范围0℃至30℃,持续10至30分钟)下,在还原剂(lialh
4
)存在条件下,将所述式i化合物在thf中进行反应,产生相应的醇;c)在第二反应条件(2至4小时)下,将步骤a)中得到的所述相应醇与炔丙基溴与氢化钠在催化剂(四丁基碘化铵(tbai))存在下在回流无水thf中进行炔化反应得到相应的炔烃衍生物;d)在第三反应条件(温度范围20℃至35℃,持续25至35小时)下,使步骤b)中得到的所述相应炔烃衍生物与叠氮化物发生铜催化的点击反应,得到所述式ii化合物。
[0051]
现在参考附图,更具体地,参考附图1至20展示了优选的实施例,类似的参考字符在整个附图中一致地表示相应的特征。
[0052]
图1展示了根据本公开所述实施例的发明的示意图。本发明涉及一种有机化学中
新开发的杂环化合物合成的工艺/方法及其抗癌、抗结核活性。实施系统性发明可以描述为以下单元本发明的元素或组分或单元:a.吡啶稠杂环新型级联合成法的发现b.取代的5-氮吲哚衍生物的合成c.取代的γ-咔啉衍生物的合成d.取代的呋喃并[b]吡啶、苯并呋喃并吡啶衍生物及呋喃[b]吡啶-靛红杂合物的合成e.光学性能评价f.抗肿瘤细胞系及抗耐多药病原体的体外研究g.新型5-氮吲哚、呋喃吡啶及其靛红杂合物的抗分枝杆菌性能h.试验流程或方法各单元功能说明:1.简要介绍了优化合成流程的意外发现和系统性开发2.本发明的合成方法和5-氮吲哚衍生物合成的底物范围、合成和区域选择性的机制说明、新型cb2激动剂类似物的合理设计与合成3.采用新开发方法合成γ-咔啉和bet抑制剂类似物4.呋喃并吡啶和苯并呋喃并吡啶的合成5.新型咔啉的光物理研究:紫外吸收和荧光研究6.典型咔啉衍生物的细胞摄取(共聚焦显微镜)和细胞毒性研究7.新型5-氮吲哚和/或呋喃并[b]吡啶及其靛红杂合物的抗分枝杆菌性能
[0053]
吡啶稠杂环新型级联合成法的发现:氮吲哚在医药产品中有着广泛的应用,是一类有效的杂环基序。其结构与有意义的生物分子结构相似,在生物系统中的可用性有限,使它们成为调节天然配体-蛋白质相互作用的首选。吡咯甘氨酸可能成为通过异环化策略开发一系列氮吲哚核心分子的关键结构成分。在尝试在回流甲苯中,在h
ü
nig碱(dipea)存在下,通过n-苄基吡咯-2-甲醛(1a)与甘氨酸甲酯盐酸盐(2a)亚胺化制备吡咯甘氨酸时,我们发现产物不是相应的亚胺,但后来被鉴定为1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3aa)。
[0054]
对一个前所未有的吡咯吡啶(氮吲哚)合成的有意义的反应进行了进一步的分析,以找出最佳反应条件。针对获得所需转化的效果研究了各种无机和有机碱(表1)。通过系统地改变n-取代吡咯-2-醛和碱的当量,找到了一个能够获得最佳产率的合适的摩尔比。考察了各种溶剂体系以提高所需5-氮吲哚的产率。
表15-氮吲哚合成反应条件的优化
[0055]
图2展示了根据本公开所述实施例,从n-取代吡咯-2-醛1a-i合成5-氮杂吲哚3aa-fa。考察了多种反应条件,包括纯反应和微波辐射。在我们手中的一系列实验数据中,我们得出结论:通过新发现的反应,在3.5当量的dipea存在下,2.0当量n-苄基吡咯-2-甲醛(1a)和1.0当量的甘氨酸烷基酯盐酸盐的混合物,在150℃下在密封管中加热,所需5-氮吲哚(3aa)的最佳产率为58%(如图2所示)。
[0056]
取代的5-氮吲哚衍生物的合成:此外,对一系列n-取代吡咯-2-醛(1b-j)反应,观察到当吡咯-2-甲醛亚基,即4-甲氧基苄基(1b)、甲基(1c)官能团,上存在富电子n-取代基时,氮吲哚的转化平稳进行。对甲苯磺酰基(1d)和苯磺酰基(1e)等具有温和拉电子基团的吡咯-2-甲醛衍生物经过类似的杂环化反应得到相应的氮吲哚衍生物,但反应时间较长且产率适中。当n-boc吡咯-2-甲醛(1h)或5h-吡咯[2,1-a]异吲哚-3-乙醛(1i)在近似的反应条件下时,由于boc保护基在1h内具有较强的吸电子倾向,并且在li中具有高应变吡咯稠化异吲哚环系统,因此没有生成得到氮吲哚产物的痕迹。综上所述,我们总结出吡咯杂环单元的c-3亲核性对杂环化起到至关重要的作用。
[0057]
图3展示了根据本公开所述实施例,1-取代-2-吡咯醛(1a-i)的1,4,5-位上的各种取代基在形成5-氮吲哚3aa-fa中的作用。吡咯亚基上的c-4取代在方法学中也是不允许的,很可能是由于空间拥挤。虽然,在c-5上对空间位阻要求较低的取代基(例如甲基基团(1f))具有良好的耐受性,但在吡咯单元的c-5位置上的大体积取代基(如苄基基团)即使在150℃
下加热48小时也不会形成5-氮杂吲哚产物。
[0058]
图4展示了根据本公开所述实施例,5-氮吲哚(3aa-fa)、咔啉(18aa-fa)和氮杂苯并呋喃(20aa-ba)和吡咯苯并呋喃(28aa-ac)的合理形成机制。各种杂环或苯并稠化杂环2-醛(1a-i、17a-h、19a-b和27)转化为相应的稠合吡啶杂环(3aa-fa、18aa-fa、20aa-ba和28aa-ac)的可能机理(如图4所示)涉及由杂环2-醛和甘氨酸烷基酯(2a-c)最初形成反式亚胺4。在hunig碱的存在下,通过从反式亚胺4中提取活性亚甲基质子而生成亚氨基亲核体5,亚氨基亲核体5与另一个杂环-2-醛分子(1a-i,17a-h,19a-b,27)进一步进行亲核加成反应,得到中间体亚氨基醇6。亚氨基醇6在反应条件下去除水分子,得到亚氨基烯胺中间体7.中间体7中的亚胺双键被共轭酸(+bh)活化,在杂环单元的3位进行亲电芳香取代,亚胺中间体8由于质子损失而芳构化,形成第二个c-c键,得到中间体10。10通过自然氧化作用的在原位脱氢生成所需的吡啶稠合化合物(3aa-fa、18aa-fa、20aa-ba和28aa-ac)。
[0059]
图5展示了根据本公开所述实施例,对各种杂环的5-区域单体而不是6-区域异构体的形成的说明。5-区域异构体的排他性形成是通过调用反式亚胺亲核剂5a参与第一个c-c键形成的过程来解释的,该过程通过向杂环-2-氨基乙醛1进行亲核加成得到5-区域异构体中间体8a,而6-区域异构体中间体8b的形成由于合成亚胺5b与杂环-2-乙醛1的亲核反应的空间拥挤而不被看好(如图5)。亚胺中间体8a在杂环或苯并稠合甲醛的3位进行亲核取代,形成第二个c-c键,通过杂环化反应闭合环,得到二氢中间体9,该中间体9在芳香化和自氧化过程中提供各种杂环,如5-氮吲哚、γ-咔啉,5-氮苯并呋喃和吡咯苯并呋喃。
[0060]
图6展示了根据本公开所述实施例,5-氮吲哚衍生物3aa(ccdc 1836867)的单晶xrd分析。进一步,酯3aa与氨水在碱性甲醇溶液中反应生成相应的酰胺衍生物11。11在磷酰氯中轻度脱水提供氰基-氮吲哚12。酯3aa与氢化铝锂快速反应生成相应的醇13。
[0061]
图7展示了根据本公开所述实施例,新型5-氮吲哚衍生物(11至14)的合成。在六元环的氮吲哚中引入甲酰基部分,保留吲哚的高度亲核的c-3和c-2位置也可以通过该方法实现,因为在回流二氯甲烷中,通过二氧化锰的处理,乙醇13可以很容易氧化成醛14(如图7所示)。
[0062]
图8展示了根据本公开所述实施例,拟定cb2激动剂(16)的合成。大麻素受体是一类跨膜蛋白,属于g蛋白偶联受体超家族。这些受体有两种类型,即cb1和cb2。大麻素-大麻素受体的相互作用因其抗姑息作用而被广泛研究,而cb2受体用于选择性靶向治疗神经病理性疼痛。近期文献表明,cb2可以成为开发新抗癌分子的重要靶点,因为cb2激动剂可以调节包括细胞存活、血管生成和转移在内的关键细胞信号通路。据推测,化合物3ca与文献报道的选择性cb2激动剂gsk554418a具有显著的结构相似性,并有望表现出类似的生物活性。因此,3ca的甲酯被水解成相应的5-氮吲哚酸(15)。此后,15与吗啉偶联得到16(如图8所示)。本实验室正在进行体外研究,以确认3ca和16在肿瘤细胞系中的cb2激动活性。
[0063]
图9展示了根据本公开所述实施例,γ-咔啉18aa-fa的合成。咔啉是嗅结构域,额外终端(bet)蛋白作为治疗癌症的一类新靶点已经出现。人类基因组编码46个含溴结构域的蛋白,大体上可分为8个亚家族。现有研究表明,通过溴结构域的选择性靶向进行的基因转录的精细调控可能为我们治疗癌症的方式带来创新性的改变。bet抑制剂正迅速进入针对各种人类癌症的临床试验,例如ibet-762(申办方gsk,用于治疗上皮组织癌症,目前处于临床试验i期)和otx015(申办方onocethix,用于治疗白血病,目前处于临床试验i期)。近年
来,一些合成的γ-咔啉衍生物在低纳摩尔浓度下表现出选择性的溴结构域抑制作用。
[0064]
取代的γ-咔啉衍生物的合成:如在提出的机制中所描述的(如图4所示),设想可以通过本发明的方法制备吡啶稠杂环库。为了验证我们的假设,我们用标准反应制备了一系列n-取代吲哚-2-甲醛衍生物(17a-h)。n-甲基吲哚-2-甲醛(2当量)和甘氨酸甲酯盐酸盐(2a-c,1当量)在密封管中用dipea(3.5当量)120℃加热3至9小时,得到相应的γ-咔啉衍生物(18aa-fa,方案6)。值得注意的是,在方案的标准反应条件下,n-保护的吲哚-2-甲醛衍生物比近期n-取代吡咯-2-甲醛衍生物(1a-i)更具活性。
[0065]
图10展示了根据本公开所述实施例,γ-咔啉衍生物18ac(ccdc:1897787)的单晶xrd分析。
[0066]
图11展示了根据本公开所述实施例,呋喃并[b]吡啶(20aa-ba)的合成。取代的呋喃并[b]吡啶、苯并呋喃并吡啶衍生物及呋喃[b]吡啶-靛红杂合物的合成呋喃甲醛(19a)和5-甲基呋喃甲醛(19b)经过实验室制定的标准反应条件,成功地将这些底物转化为相应的呋喃并[b]吡啶(20aa-ba)。发现这类化合物的合成衍生物针对引发结核病的分枝杆菌菌株表现出抗结核特性。此外,合成了呋喃并[b]吡啶-靛红杂合物分子(26),以进一步提高抗结核活性。
[0067]
图12展示了根据本公开所述实施例,用于抗结核活性的呋喃吡啶-靛红杂合物(26)的合成。通过新型呋喃并[b]吡啶的初步评价发现其十分具有前途的。呋喃并[b]吡啶20ba经lialh4处理得到相应的醇21。该醇经炔丙基化反应得到炔衍生物22,合成叠氮靛25,并根据文献报道合成了靛红-叠氮化物25。22和25的所述点击化学反应顺利转化为新型呋喃并吡啶-靛红杂合物26(如图10所示)。对结核分枝杆菌的耐多药和广谱耐药(mdr)株中所有呋喃并吡啶衍生物和靛红杂合物进行了抗分枝杆菌筛选,并对一组生物膜形成菌或病原体进行了抗生物膜活性筛选,发现其具有意义的抗结核活性。
[0068]
图13展示了根据本公开所述实施例,苯并呋喃并吡啶(28aa-ac)的合成。通过苯并呋喃-2-甲醛27与甘氨酸烷基酯(2a-c)反应以获得新的苯并呋喃稠吡啶衍生物(28aa-ac),再次证明了发现方法的多功能性,如图11所示。用本方法合成的所有杂环是它们各自反应的唯一可识别产物。
[0069]
图14展示了根据本公开所述实施例,苯并呋喃并吡啶(28aa-ac)的合成。光学性能评价:对合成的一种咔啉衍生物18ac的光物理性质进行了研究。在四种不同的有机溶剂,即二氯甲烷(ch
2
cl
2
)、甲醇(meoh)、二甲基亚砜(dmso)和正己烷中制备了10至5m的18ac溶液。发现化合物18ac的紫外吸收特性与溶剂极性无关。在230nm处观察到18ac的dmso溶液的最大吸收峰(λmax)。随后,用同样四种溶液对18ac进行了荧光研究,当溶剂由非极性己烷改为中极性二氯甲烷,再改为高极性dmso时,可观察到近40nm的红移(表2;图14)。18ac的固有荧光在极性质子溶剂甲醇中被猝灭,这可能是由于在高极性溶剂中氢键相互作用促进的超快溶质-溶剂分子间光致电子转移。
表218ac的吸产率(ε)和半衰期(τ)的测量
[0070]
图15展示了根据本公开所述实施例,18ac在dmso中(左侧;λexc 360nm)和10至5m化合物18ac溶液在四种不同溶剂中在紫外室内(右侧)的荧光衰减曲线。采用时间相关单光子计数(tcspc)实验测定荧光寿命。一般来说,荧光寿命越长,量子产率越高,成像对比度越好。有机分子是fret研究的理想选择,因为它们被归为“刚性染料”。令人高兴的是,18ac在dmso和dcm中具有很强的荧光,平均荧光寿命分别为8.35纳秒(ns)和4.73纳秒(表2;图15)。
[0071]
图16显示了在hela、mcf-7、hek293、a431和a549细胞系中,咔啉18ac、18bc、18da和18fa孵育48小时的剂量-反应曲线。根据本公开所述实施例,通过一式三份的标准结晶紫分析测定ic50值;体外研究:采用标准结晶紫法对人恶性细胞系,例如hela、mcf-7、hek293、a431和a549,对新型咔啉衍生物18ac、18bc、18da和18fa进行细胞毒性试验。将细胞种板于96孔板(每孔1800个细胞),并在37℃、5%co
2
条件下培养48小时。达到50%的融合度时,分别用一系列浓度(0.1μm、0.25μm、0.5μm、1μm、2.5μm、5μm、10μm、25μm、50μm和100μm)的咔啉18ac、18bc、18da和18fa孵育细胞48小时。这些检测的所得结果(表3)显示,微摩尔浓度的咔啉具有细胞毒性。表3咔啉18a、18bc、18da和18fa作为dna嵌入剂在hela、mcf7、hek293、a431和a549细胞中的ic
50
研究(培养48小时)。
[0072]
图17展示了根据本公开所述实施例,用于hela细胞中18ac摄取的共焦显微镜研究(λex=405nm;收集范围=420至470nm)。a)10μm浓度的18ac孵育3h后hela细胞的共聚焦荧光图像(20倍放大,2倍变焦);b)hela细胞的dic图像c)(a)和(b)重叠图d)100nm浓度的18ac孵育3h后hela细胞的共聚焦图像(20倍放大,2倍变焦)e)hela细胞的dic图像f)(d)和(e)重叠图
[0073]
为了进一步评估新型咔啉的细胞摄入,进行了活体细胞成像实验。将细胞种板于4孔板(细胞计数=每孔10
4
个细胞),并在37℃、5%co
2
条件下培养48小时。用18ac(10nm、100nm、1μm、10μm和100μm)孵育3小时后,用共聚焦显微镜(λex=405nm;收集范围=420至470nm)监测细胞摄入和分布。用10μm的浓度孵育时,在癌细胞中观察到18ac的适当胞质摄
入量,而在100nm浓度下观察到标称摄入量。
[0074]
图18展示了根据本公开所述实施例,各种化合物的结构式。
[0075]
图19展示了根据本公开所述实施例,将本发明与现有方法区分开的本发明的关键特征。
[0076]
新型呋喃并[b]吡啶及其靛红杂合物对非致病性(耻垢分枝杆菌)和致病性/机会致病性分枝杆菌(牛分枝杆菌)的体外抗分枝杆菌活性
[0077]
新合成的呋喃并[b]吡啶-靛红杂合物衍生物26(也被标记为cv-pd-pf-ist-hbd-001)对非致病性(耻垢分枝杆菌)和bcg致病性(牛分枝杆菌)耐多药株进行了检测,结果如图20所示。观察到呋喃并[b]吡啶-靛红杂合物药26在70至100μm的浓度范围内分别对非致病性分枝杆菌株在24小时内具有活性,对致病性耐多药分枝杆菌株在48小时内具有活性。所得结果令人兴奋,并加强了药物设计中的修改或改变,以便由印度开发新的抗结核药物,以结束发展中国家数百万人的死亡。
[0078]
图20展示了根据本公开所述实施例,呋喃并[b]吡啶-靛红杂合物衍生物26针对耻垢分枝杆菌(50至100μm)的体外抗结核活性,sd(n=3);耐多药/机会致病性牛分枝杆菌(70至100μm)sd(n=3)。试验方法:
[0079]
癌细胞系中咔啉的细胞毒性试验方法:为此,对咔啉18ac、18bc、18da和18fa进行了标准细胞活性研究(结晶紫试验),以检测对各种人肿瘤细胞系,例如hela、mcf-7、hek293、a431和a549,的抗癌活性。结晶紫是一种三芳基甲烷染料,通过与dna中的核糖部分结合来染色粘附细胞。实验中结晶紫染色的量与反复洗涤后附着在平板上的活细胞生物量成正比。将上述人癌细胞系种板于96孔组织培养板(1800个细胞/孔)中,用rpmi 1640培养基(体积≥100μl/孔)在37℃、5%co
2
条件下培养48小时,添加10%热灭活胎牛血清(hifbs)和1%青霉素-链霉素抗生素。当单层细胞达到50%的融合度/孔后,吸出旧培养基并替换为≥100μl/孔的新鲜培养基,并分别加入增加浓度的咔啉18ac、18bc、18da和18fa(0.1μm、0.25μm、0.5μm、1μm、2.5μm、5μm、10μm、25μm、50μm和100μm),并在37℃、5%co
2
条件下培养48小时。阳性对照:将40%dmso的培养基加入到三个孔中,在相同条件下培养。每种浓度处理三孔细胞。在48小时后吸出培养基/孔,用温和的自来水流冲洗细胞两次,并将板倒置在滤纸上以除去残余液体。为了测量细胞活力,将50μl的0.5%结晶紫染色溶液(0.5g结晶紫粉末,80mlh
2
o,20ml甲醇)添加到每个孔中,并在室温下在摇床上孵育20分钟(频率约为20次震荡/分钟)。如前所述,再次轻轻冲洗板,并在室温下晾干2小时。然后通过添加200μl甲醇/孔来溶解结晶紫染料,并在室温下孵育板20分钟。最后,使用synergy h1多模式读板机(biotek instruments,inc.,winooski,vt,usa)在570nm(od570)处测量每个孔的光密度。测量空白孔od570的平均背景,并从板的od570/孔中减去该平均值。测定处理后的活细胞百分比,并使用graphpad prism v.6.02(graphpad软件,san diego,ca)绘制剂量-反应曲线作为半对数[浓度]与标准化细胞活力百分比曲线。计算了咔啉18ac、18bc、18da和18fa对不同人肿瘤细胞系(图16)的半数抑制浓度(ic50),如表3所示。
[0080]
图17展示了根据本公开所述实施例,用于hela细胞中18ac摄取的共焦显微镜研究(λex=405nm;收集范围=420至470nm);
[0081]
图18展示了根据本公开所述实施例,各种化合物的结构式;
[0082]
图19展示了根据本公开所述实施例,将本发明与现有方法区分开的本发明的关键特征。
[0083]
分枝杆菌株的细菌培养制备及体外灭活试验方法细菌培养制备i.在含有吐温80的7h9 middlebrook培养基中接种单克隆耻垢分枝杆菌(m.smeg)或牛分枝杆菌(bcg)ii.在37℃条件下进行细菌培养,持续36至48小时iii.在室温(rt)下,取1ml生长的耻垢分枝杆菌或牛分枝杆菌(bcg),以5000rpm的转速离心5分钟。iv.去除上清液,将沉淀溶解在1ml高压灭菌的1x pbs中。v.同样,培养物在室温下以5000rpm离心5分钟vi.将此时形成的沉淀溶解于1x pbs以进行下一步试验分枝杆菌体外灭活试验a.测量耻垢分枝杆菌或牛分枝杆菌的od
600
,并设置为0.1b.在微量离心管中单独制备细菌和不同药物浓度(5-氮吲哚或呋喃并[b]吡啶或呋喃并[b]吡啶-靛红杂合物)的混合物(所有浓度的混合物体积保持在200μl)c.含有细菌和药物混合物的试管在37℃条件下培养0、6、12、24小时d.在每个单独的时间点之后,为含所有浓度的每组制备高达10-5
的1:10梯度稀释液(使用1
×
pbst)e.将10-3
、10-4
、10-5
(5μl斑点)的板放入含有7h9/lb琼脂培养基的培养皿中f.将板在37℃条件下培养36小时或更长时间,以便出现肉眼可见的克隆菌落g.使用菌落计数器进行菌落计数
[0084]
图20展示了根据本公开所述实施例,呋喃并[b]吡啶-靛红杂合物衍生物26针对耻垢分枝杆菌(50至100μm)的体外抗结核活性,sd(n=3);耐多药/机会致病性牛分枝杆菌(70至100μm)sd(n=3)。。示例
[0085]
示例1:合成稠合吡啶杂环(3aa-fa、18aa-fa、20aa-ba和28aa-ac)的一般方法。将醛(1a-i、17a-h、19a-b或27,2.00mmol)、甘氨酸烷基酯盐酸盐(2a-c,1.00mmol)与n,n-二异丙基乙胺(dipea,3.50mmol)的混合物在120至150℃下在密封管(25ml,硼硅酸盐)中加热3至12小时,并持续搅拌(通过tlc监测)。将反应混合物冷却至室温,用ch
2
cl
2
(1
×
10ml)稀释并用盐水(1
×
10ml)洗涤。将反应混合物用ch
2
cl
2
(3
×
10ml)进一步萃取。将合并的有机层在无水na
2
so
4
上干燥,过滤,浓缩,并将己烷-etoac剂混合物作为洗脱剂来通过中性矾土(175目)柱层析法进行纯化。
[0086]
示例2:1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3aa)。根据上述一般方法,,将1a(100mg,0.54mmol)、2a(34mg,0.27mmol)和dipea(0.165ml,0.95mmol)在密封管中150℃加热6小时。在处理后,将正己烷-etoac(94:6)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率58%(66mg);黄色晶状固体;熔点=134至136℃;rf0.65(2:1己烷-etoac);ir(kbr)3028(=c-h),2922-2850(c-h),1722(c
=o),1712-1554(c=c),1357(c-h弯曲),779(=c-h弯曲)cm-1;1h nmr(500mhz,cdcl
3
)δ8.01(s,1h),7.35-7.27(m,3h),7.24(d,j=3.2hz,1h),7.18-7.13(m,2h),7.12-7.08(m,3h),7.07-7.02(m,2h),6.91(d,j=3.2hz,1h),6.90-6.87(m,1h),6.82(dd,j=3.7,1.7hz,1h),6.29(dd,j=2.9,2.3hz,1h),5.88(s,2h),5.34(s,2h),3.95(s,3h);
13
c nmr(100mhz,cdcl
3
)δ167.4,145.8,140.5,139.6,138.7,136.2,131.2,130.4,129.0,128.2,128.2,127.1,127.0,126.8,125.7,125.0,113.2,108.2,105.7,103.7,52.5,51.7,50.2;hrms(esi)[c
27
h
23
n
3
o
2
h
+
]计算值422.1863,实测值422.1859。
[0087]
示例3:1-(4-甲氧基苄基)-4-(1-(4-甲氧基苄基)-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3ba)。根据上述一般方法,,将1b(100mg,0.46mmol)、2a(29mg,0.23mmol)和dipea(0.140ml,0.81mmol)在密封管中于150℃加热6小时。在处理后,将己烷-etoac(88:12)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率61%(68mg);黄色液体;rf 0.60(1:1己烷-etoac);ir(kbr)3073(=c-h),2958-2851(c-h),1743(c=o),1109-1029(c-o)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.04(s,1h),7.22(d,j=3.2hz,1h),7.08(d,j=8.5hz,2h),7.02(d,j=8.5hz,2h),6.90-6.81(m,4h),6.78(dd,j=3.5,1.5hz,1h),6.69(d,j=8.8hz,2h),6.25(dd,j=3.3,2.6hz,1h),5.77(s,2h),5.28(s,2h),3.97(s,3h),3.78(s,3h),3.69(s,3h);
13
c nmr(100mhz,cdcl
3
)δ166.7,158.8,157.8,145.1,139.6,137.9,130.9,130.3,129.6,127.9,127.8,127.4,124.7,124.4,113.7,112.9,112.5,107.3,105.0,102.8,54.6,54.4,51.7,50.4,49.0;计算[c
29
h
27
n
4
o
3
+h
+
]482.2074的hrms(esi),等于482.2074.
[0088]
示例4:1-甲基-4-(1-甲基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3ca)。根据上述一般方法,,将1c(100mg,0.92mmol)、2a(29mg,0.46mmol)和dipea(0.281ml,1.61mmol)在密封管中于150℃加热6小时。在处理后,将己烷-etoac(92:8)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率48%(59mg);黄褐色液体;rf 0.60(1:1己烷-etoac);ir(kbr)3126-3084(=c-h),2926-2852(c-h),1732(c=o),1714-1556(c=c),1350(c-h bend),721(=c-h bend)cm-1;
1
h nmr(400mhz,cdcl3)δ8.04(s,1h),7.24-7.17(m,1h),6.89-6.81(m,1h),6.80-6.74(m,1h),6.73-6.68(m,1h),6.25-6.13(m,1h),4.03(s,3h),3.97(s,3h),3.85(s,3h);13c nmr(100mhz,cdcl
3
)δ167.5,145.7,140.7,138.5,132.0,130.7,126.2,124.9,112.6,107.5,105.5,103.1,52.5,36.4,33.1;hrms(esi)[c
15
h
15
n
3
o
2
+h
+
]计算值270.1237,实测值270.1233。
[0089]
示例5:1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸叔丁酯(3ac)。根据上述一般方法,,将1a(100mg,0.54mmol)、2c(45mg,0.27mmol)和dipea(0.165ml,1.61mmol)在密封管中于150℃加热8小时。在处理后,将己烷-etoac(95:5)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率40%(50mg);黄色液体;rf 0.50(4:1己烷-etoac);ir(kbr)3063(=c-h),2976-2849(c-h),1732(c=o),1701-1564(c=c),1363(c-h弯曲),723(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ7.93(s,1h),7.37-7.26(m,3h),7.22(d,j=3.2hz,1h),7.19-7.04(m,7h),6.91(d,j=3.2hz,1h),6.89-6.82(m,2h),6.26(dd,j=3.2,3.0hz,1h),6.03(s,2h),5.33(s,2h),1.59(s,9h);
13
c nmr(100mhz,cdcl3)δ166.0,145.4,140.7,140.2,139.8,136.3,130.9,130.5,129.0,128.3,128.1,127.2,127.0,126.8,125.8,124.3,113.3,108.1,105.0,103.5,81.1,51.8,50.1,
28.3;hrms(esi)[c
30
h
29
n
3
o
2
+h
+
]计算值464.2333,实测值464.2334.
[0090]
示例6:1-甲基-4-(1-甲基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸乙酯(3cb)。根据上述一般方法,,将1c(100mg,0.92mmol)、2b(64mg,0.46mmol)和dipea(0.281ml,1.61mmol)在密封管中于150℃加热7小时。在处理后,将己烷-etoac(92:8)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率46%(60mg);淡黄色液体;rf 0.50(2:1己烷-etoac);ir(kbr)3077(=c-h),2957-2850(c-h),1731(c=o),1714-1558(c=c),1374(c-h弯曲),725(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.04(s,1h),7.24-7.19(m,1h),6.92-6.85(m,1h),6.83-6.78(m,1h),6.78-6.72(m,1h),6.27-6.19(m,1h),4.46(q,j=6.8hz,2h),4.10(s,3h),3.88(s,3h),1.45(t,j=6.8hz,3h);
13
c nmr(100mhz,cdcl
3
)δ166.9,145.5,140.7,138.7,131.9,130.7,126.3,124.6,112.7,107.5,105.2,103.0,61.3,36.6,33.1,14.4;hrms(esi)[c
16
h
17
n
3
o
2
+h
+
]计算值284.1394,实测值284.1389。
[0091]
示例7:1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸乙酯(3ab)。根据上述一般方法,,将1a(100mg,0.54mmol)、2b(38mg,0.27mmol)和dipea(0.165ml,0.95mmol)在密封管中于150℃加热6小时。在处理后,将正己烷-etoac(94:6)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率55%(65mg);黄色液体;rf 0.60(4:1己烷-etoac);ir(kbr)3056(=c-h),2977-2851(c-h),1729(c=o),1712-1554(c=c),1367(c-h弯曲),726(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ7.99(s,1h),7.38-7.26(m,3h),7.26-7.21(m,1h),7.19-7.13(m,2h),7.13-7.02(m,5h),6.95-6.90(m,1h),6.90-6.86(m,1h),6.85-6.80(m,1h),6.33-6.24(m,1h),5.94(s,2h),5.35(s,2h),4.41(q,j=6.8hz,2h),1.38(t,j=6.8hz,3h);
13
c nmr(100mhz,cdcl
3
)δ166.9,145.7,140.6,139.7,139.0,136.3,131.1,130.4,129.0,128.3,128.2,127.2,127.0,126.8,125.8,124.8,113.3,108.2,105.5,103.6,61.3,51.7,50.2,14.4;hrms(esi)[c
28
h
25
n
3
o
2
+h+]计算值436.2020,实测值436.2024。
[0092]
示例8:1-(4-甲氧基苄基)-4-(1-(4-甲氧基苄基)-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸乙酯(3bb)。根据上述一般方法,,将1b(100mg,0.46mmol)、2a(32mg,0.23mmol)和dipea(0.140ml,0.81mmol)在密封管中于150℃加热8小时。在处理后,将己烷-etoac(90:10)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率60%(68mg);黄色液体;rf 0.55(2:1己烷-etoac);ir(kbr)3067(=c-h),2955-2852(c-h),1738(c=o),1713-1515(c=c),1369(c-h弯曲),1106-1028(c-o),727(=c-h弯曲)cm-1;1h nmr(400mhz,cdcl3)δ8.03(s,1h),7.21(d,j=3hz,1h),7.08(d,j=8.5hz,2h),7.05(d,j=8.8hz,2h),6.88(d,j=3.0hz,1h),6.87-6.82(m,3h),6.80(dd,j=3.3,1.2hz,1h),6.69(d,j=8.8hz,2h),6.26(dd,j=3.0,2.5hz,1h),5.83(s,2h),5.28(s,2h),4.43(q,j=7.0hz,2h),3.78(s,3h),3.70(s,3h),1.40(t,j=7.0hz,3h);
13
c nmr(100mhz,cdcl
3
)δ167.0,159.5,158.5,145.7,140.4,138.9,131.7,130.9,130.3,128.6,128.6,128.2,125.4,124.8,114.4,113.7,113.2,108.0,105.5,103.5,61.3,55.3,55.2,51.1,49.7,14.4;hrms(esi)[c
30
h
29
n
3
o
4
+h
+
]计算值496.2231,实测值496.2230。
[0093]
示例9:1-(4-甲氧基苄基)-4-(1-(4-甲氧基苄基)-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸叔丁酯(3bc)。根据上述一般方法,,将1b(100mg,0.46mmol)、2c(39mg,
0.23mmol)和dipea(0.140ml,0.81mmol)在密封管中于150℃加热7小时。在处理后,将己烷-etoac(9:1)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率42%(50mg);黄橙色油状液体;rf 0.55(2:1己烷-etoac);ir(kbr)3066(=c-h),2995-2833(c-h),1730(c=o),1715-1554(c=c),1366(c-h弯曲),1113-1033(c-o),727(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ7.96(s,1h),7.19(d,j=3.3hz,1h),7.08(d,j=8.6hz,2h),7.04(d,j=8.5hz,2h),6.88(d,j=3.3hz,1h),6.87-6.79(m,4h),6.69(d,j=8.5hz,2h),6.25(dd,j=3.5,2.4hz,1h),5.93(s,2h),5.26(s,2h),3.78(s,3h),3.69(s,3h),1.61(s,9h);
13
c nmr(100mhz,cdcl3)δ166.1,159.5,158.5,145.4,140.6,140.1,131.8,130.7,130.4,128.7,128.6,128.3,125.5,124.3,114.4,113.7,113.3,108.0,105.0,103.4,81.1,55.3,55.2,51.3,49.7,28.3;hrms(esi)[c
32
h
33
n
3
o
4
+h
+
]计算值524.2544,实测值524.2551。
[0094]
示例10:1-对甲苯磺酰基-4-(1-对甲苯磺酰基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3da)。根据上述一般方法,,将1d(100mg,0.40mmol)、2a(25mg,0.20mmol)和dipea(0.122ml,0.70mmol)在密封管中150℃加热12小时。在处理后,将己烷-etoac(85:15)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率47%(52mg);灰白色固体;熔点=120至122℃;rf 0.50(1:1己烷-etoac);ir(kbr)3132-3064(=c-h),2955-2850(c-h),1728(c=o),1710-1512(c=c),1371(c-h弯曲),1309(n-s=o),1145(s=o),725(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl3)δ8.72(s,1h),7.93(d,j=8.0hz,2h),7.86(d,j=8.3hz,2h),7.70(d,j=3.8hz,1h),7.41-7.36(m,1h),7.32(d,j=8.0hz,2h),7.29-7.25(m,2h),6.66(d,j=3.8hz,1h),6.41-6.36(m,1h),6.33(dd,j=3.3,2.6hz,1h),4.03(s,3h),2.40(s,3h),2.38(s,3h);
13
c nmr(100mhz,cdcl
3
)δ166.1,146.2,145.5,145.0,142.0,139.3,136.0,134.8,131.0,130.5,130.2,129.8,129.3,128.3,127.2,124.3,117.3,112.2,110.2,108.1,53.0,29.8,21.7;hrms(esi)[c
27
h
23
n
3
o
6
s
2
+h
+
]计算值550.1101,实测值550.1096。
[0095]
示例11:1-(苯磺酰基)-4-(1-(苯磺酰基)-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3ea)。根据上述一般方法,,将1e(100mg,0.43mmol)、2a(27mg,0.21mmol)和dipea(0.128ml,0.74mmol)在密封管中150℃加热12小时。在处理后,将己烷-etoac(85:15)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率45%(49mg);黄色固体;熔点=104至106℃;rf0.50(1:1己烷-etoac);ir(kbr)3132-3064(=c-h),3005-2850(c-h),1728(c=o),1710-1512(c=c),1371(c-h弯曲),1309(n-s=o),1145(s=o),725(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.74(s,1h),8.06(d,j=7.5hz,2h),7.99(d,j=7.5hz,2h),7.71(d,j=3.8hz,1h),7.67-7.43(m,6h),7.40(dd,j=3.0,1.5hz,1h),6.67(d,j=3.8hz,1h),6.42(dd,j=3.0,1.5hz,1h),6.35(dd,j=3.3,2.5hz,1h),4.01(s,3h);
13
c nmr(100mhz,cdcl
3
)δ166.0,145.3,142.1,139.3,139.0,137.7,134.8,133.8,131.0,129.8,129.6,129.3,129.1,128.1,127.1,124.4,117.5,112.3,110.1,108.2,52.9;hrms(esi)c
25
h
19
n
3
o
6
s
2
+na+]计算值544.0607,实测值544.0603。
[0096]
示例12:1-苄基-4-(1-苄基-5-甲基-1h-吡咯-2-基)-2-甲基-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3fa)。根据上述一般方法,,将1f(100mg,0.50mmol)、2a(32mg,0.25mmol)和dipea(0.150ml,0.88mmol)在密封管中于150℃加热6小时。在处理后,将己烷-etoac(90:10)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗残渣;产率64%(72mg);黄色液
体;rf 0.55(2:1己烷-etoac);ir(kbr)3027(=c-h),2949-2852(c-h),1727(c=o),1712-1539(c=c),1355(c-h弯曲),782(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl3)δ7.88(s,1h),7.43-7.26(m,3h),7.19-7.01(m,3h),6.99-6.90(m,2h),6.89-6.82(m,2h),6.80-6.66(m,2h),6.12-6.02(m,1h),5.92(s,2h),5.33(s,2h),3.87(s,3h),2.38(s,3h),2.22(s,3h);
13
c nmr(100mhz,cdcl3)δ
13
c nmr(100mhz,cdcl3)δ167.5,144.7,141.4,140.5,139.9,138.0,136.5,133.2,130.4,129.5,129.0,128.2,127.7,126.3,126.1,126.0,112.3,107.5,105.1,102.5,52.3,47.8,46.8,12.9,12.7;hrms(esi)c
29
h
27
n
3
o
2
+h
+
]计算值450.2176,实测值450.2173。
[0097]
示例13:1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-甲酰胺(11)。在圆底烧瓶(100ml)中,将1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3aa,100mg,0.24mmol)和koh(14mg,0.24mmol)溶解在甲醇(5ml)中,在室温下连续搅拌。使用玻璃注射器将氨水(25%,0.350ml,9.40mmol)逐滴添加到混合物中,持续10分钟。在室温下将反应混合物继续搅拌24小时。反应完成后,在减压条件下蒸发meoh。向残渣中加入milliq水(5ml)和etoac(5ml),分离有机层。用etoac(5
×
3ml)进一步萃取水相。将合并的有机层在无水na
2
so
4
上干燥,过滤,并在减压条件下浓缩。以etoac为洗脱液,采用中性矾土(175目)柱层析法纯化残渣;产率78%(150mg),黄色胶状液;rf 0.50(etoac);ir(kbr)3431(n-h),3033(=c-h),2960-2852(c-h),1677(c=o),1562(c-n弯曲),1376-1360(c-h弯曲),1296-1029(c-o),726(=c-h bend)cm-1;
1
h nmr(400mhz,cdcl3)δ8.07(s,1h),7.38-7.19(m,7h),7.16-7.06(m,3h),6.95(d,j=3.0hz,1h),6.92-6.85(m,2h),6.85-6.78(m,1h),6.40(dd,j=3.0,2.6hz,1h),5.59(s,2h),5.38(s,2h),4.89(brs,2h);
13
c nmr(100mhz,cdcl3)δ168.1,144.1,141.1,140.0,136.3,131.0,129.0,129.0,128.7,128.1,127.0,127.0,126.9,126.1,125.3,124.9,112.6,108.8,103.5,103.0,51.3,50.2;ms(esi)[c
26
h
22
n
4o
+h
+
]计算值407.1866,实测值407.2023。
*
[0098]
*
化合物11在极性溶剂中不稳定,无法记录良好的hrms。
[0099]
示例14:1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-碳腈(12)。将1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-甲酰胺(11,40mg,0.098mmol)装入烘箱干燥的单颈圆底烧瓶(25ml),并使用玻璃注射器在室温下逐滴加入pocl
3
(5ml)。将回流冷凝器固定在圆底烧瓶上,将反应混合物在60℃条件下加热并搅拌过夜。反应完成后(由tlc监控),用甲苯(5ml)稀释反应混合物,并在减压条件下蒸发溶剂。将饱和nahco
3
(10ml)缓慢添加到反应混合物中,中和过量的氧氯化磷。用etoac(5
×
3ml)萃取水相。将合并的有机层在无水na
2
so
4
上干燥,过滤,并在减压条件下浓缩。以己烷-etoac(90:10)为洗脱液,采用中性矾土(175目)柱层析法纯化残渣得到12;产率66%(25mg),无色液体;rf 0.20(2:1己烷-etoac);ir(kbr)3431(n-h),3031(=c-h),2960-2852(c-h),2223(-c≡n延展),1588-1530(c=c),1376-1360(c-h弯曲),725(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ7.43(s,1h),7.39-7.31(m,3h),7.30(d,j=3.0hz,1h),7.22-7.02(m,7h),6.94(d,j=3.0hz,1h),6.93-6.89(m,1h),6.85(dd,j=3.5,1.2hz,1h),6.31(dd,j=3.0,2.4hz,1h),5.75(s,2h),5.31(s,2h);
13
c nmr(100mhz,cdcl3)δ147.3,139.2,135.5,131.7,129.5,129.2,128.5,128.5,128.4,127.2,127.0,126.9,126.6,124.6,123.1,119.2,114.1,109.1,108.4,104.1,52.1,50.6;hrms(esi)[c
26
h
20
n
4
+h
+
]计算值389.1761,实测值
389.1777。
[0100]
示例15:1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-基)甲醇(13)。在双颈圆底烧瓶(50ml)中,在惰性气氛下将1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3aa,250mg,0.59mmol)溶解在干燥thf(5ml)中。在单份添加固体lialh4(68mg,1.78mmol)之前,将反应混合物冷却至0℃。将反应混合物加热至室温并进一步搅拌20分钟。在消耗酯3aa后,如tlc所证实,用饱和nh
4
cl(10ml)溶液淬火反应混合物,并用etoac(10ml)进一步稀释。用etoac(10
×
3ml)萃取水层。将合并的有机提取物在无水na
2
so
4
上干燥,过滤,在减压条件下蒸发,使用己烷-etoac(75:25)用作洗脱液来采用中性矾土(175目)柱色谱法纯化粗残渣;产率94%(220mg);无色液体;rf 0.35(2:1己烷-etoac);ir(kbr)3414(o-h),3028(=c-h),2958-2850(c-h),1695-1559(c=c),1357(c-h弯曲),1100-1023(c-o延展)cm-1;
1
h nmr(400mhz,cdcl3)δ7.36-7.17(m,6h),7.13(d,j=3.3hz,1h),7.07(dd,j=7.5,8.0hz,4h),6.90(s,1h),6.89-6.80(m,3h),6.36(dd,j=3.0,2.6hz,1h),5.65(s,2h),5.28(s,2h),4.63(s,2h),3.24(brs,1h);
13
c nmr(100mhz,cdcl3)δ149.6,144.7,141.6,139.7,136.6,131.1,129.2,129.0,128.5,128.0,127.0,126.8,126.3,125.2,122.7,112.6,108.5,103.0,99.0,64.7,51.7,50.1;hrms(esi)[c
26
h
23
n
3
o+h
+
]计算值394.1914,实测值394.1913。
[0101]
示例16:1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-乙醛(14)。在圆底烧瓶(100ml)中,制备溶于二氯甲烷(5ml)的(1-苄基-4-(1-苄基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-基)甲醇(13,100mg,0.25mmol)溶液,并且单份添加固体mno
2
(326mg,3.75mmol)。将双层回流冷凝器固定在圆底烧瓶上,反应混合物回流过夜。反应完成后,溶剂在减压条件下蒸发。以己烷-etoac(95:5)为洗脱液,采用中性矾土(175目)柱层析法纯化残渣;产率70%(70mg);灰白色液体;rf 0.55(2:1己烷-etoac);ir(kbr)3030(=c-h),2960-2852(c-h),1696(c=o),1606-1556(c=c),1358-1331(c-h弯曲),1287-1079(c-o),725(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl3)δ10.02(s,1h),7.83(s,1h),7.42-7.27(m,4h),*7.23-7.02(m,7h),6.98-6.93(m,1h),6.92-6.88(m,1h),6.88-6.82(m,1h),6.42-6.28(m,1h),5.81(s,2h),5.36(s,2h);
13
c nmr(100mhz,cdcl3)δ194.3,146.3,145.1,140.4,139.5,136.0,132.3,130.3,129.1,128.4,128.3,127.0*,126.8,126.2,125.9,113.3,108.5,103.7,102.2,51.9,50.4;hrms(esi)[c
26
h
21
n
3
o+h
+
]计算值392.1757,实测值392.1756。
*
高强度碳
[0102]
示例17:1-甲基-4-(1-甲基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸(15)。在圆底烧瓶(50ml)中制备溶于thf(3ml)的1-甲基-4-(1-甲基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-羧酸甲酯(3ca,200mg,0.74mmol)溶液。在室温下添加1m的lioh水溶液(2.5ml),并在相同温度下搅拌反应混合物3小时(通过tlc监测)。消耗3ca后,向反应混合物中添加乙醚(10ml)和饱和nahco
3
(10ml)。分离水层并酸化至ph 4(滴加6n的hcl)。用etoac(5
×
10ml)萃取水相。在无水na
2
so
4
上干燥合并的有机层,过滤,减压条件下蒸发得到粗产物15,该产物无需进一步纯化即可在下一步骤中使用;粗产率90%(172mg),黄色油状液体;rf 0.10(etoac);
1
h nmr(400mhz,cdcl3)δ8.14(s,1h),7.32(d,j=3.0hz,1h),6.90(d,j=3.0hz,1h),6.86(m,1h),6.77(dd,j=3.5,1.3hz,1h),6.29(dd,j=2.8,2.4hz,1h),3.94(s,3h),3.91(s,3h);
13
c nmr(100mhz,cdcl3)δ164.8,142.8,140.3,135.8,132.0,128.2,
125.8,124.5,112.6,107.3,102.9,102.7,35.3,32.3;hrms(esi)[c
14
h
13
n
3
o
2
+h
+
]计算值256.1081,实测值256.1066。
[0103]
示例18:(1-甲基-4-(1-甲基-1h-吡咯-2-基)-1h-吡咯[3,2-c]吡啶-6-基)(吗啉代)甲酮(16)。将溶解于干燥dmf(4ml)的未纯化15(100mg,0.39mmol)在惰性气氛下装入双颈圆底烧瓶(50ml)。将反应混合物短暂冷却至0℃,然后在持续搅拌条件下依次添加吗啉(0.13ml,1.57mmol)、1-乙基-3-(3-二甲基氨基丙基)碳二亚胺(300mg,1.57mmol)、羟基苯并三唑(212mg,1.57mmol)和dipea(0.54ml,3.13mmol)。将反应混合物加热至室温,再次搅拌16小时。在消耗15后(经tlc确认),向反应混合物中添加冷盐水(10ml)。用etoac(10
×
3ml)萃取反应混合物,将合并的有机层在无水na
2
so
4
上干燥,过滤,浓缩,并以己烷-etoac(50:50)用作洗脱液来采用中性矾土(175目)柱色谱法进行纯化;产率49%(60mg);白色结晶固体;熔点=95至96℃;rf 0.40(1:1己烷-etoac);ir(kbr)3065(=c-h),2957-2850(c-h),1682(c=o),1641-1513(c=c),1371(c-h弯曲),723(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ7.65(s,1h),7.17(d,j=3.0hz,1h),6.82(d,j=3.0hz,1h),6.80-6.77(m,1h),6.76-6.71(m,1h),6.25(dd,j=2.8,2.4hz,1h),3.97(s,3h),3.92-3.76(m,9h),3.72-3.60(m,2h);
13
c nmr(100mhz,cdcl3)δ169.2,144.2,144.1,141.1,131.1,130.9,125.9,123.4,112.6,107.6,104.3,102.8,67.3,67.0,48.1,43.1,36.6,32.9;hrms(esi)c
18
h
20
n
4
o
2
+h+]计算值325.1659,实测值325.1655。
[0104]
示例19:5-甲基-1-(1-甲基-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸甲酯(18aa)。根据上述一般方法,,将17a(100mg,0.62mmol)、2a(39mg,0.31mmol)和dipea(0.190ml,1.09mmol)在密封管中120℃加热6小时。在处理后,将己烷-etoac(80:20)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率70%(80mg);黄色固体;熔点=210至212℃;rf 0.35(2:1己烷-etoac);ir(kbr)3055(=c-h),2956-2854(c-h),1734(c=o),1687-1534(c=c),1407-1376(c-h弯曲),782(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.31(s,1h),7.82(d,j=8.0hz,1h),7.74(d,j=8.0hz,1h),7.56(dd,j=8.0,7.3hz,1h),7.51(d,j=8.3hz,1h),7.45(d,j=8.3hz,1h),7.33(dd,j=8.0,7.8hz,1h),7.19(dd,j=7.5,7.5hz,1h),7.15(dd,j=7.5,7.6hz,1h),6.99(s,1h),4.05(s,3h),4.00(s,3h),3.75(s,3h);
13
c nmr(100mhz,cdcl
3
)δ166.9,146.3,145.8,142.9,142.2,138.3,137.6,128.1,127.9,123.1,122.4,121.3,121.2,121.1,120.8,119.8,109.8,109.1,105.7,104.3,53.0,31.0,29.6;hrms(esi)c
23
h
19
n
3
o
2
+h+]计算值370.1550,实测值370.1515。
[0105]
示例20:5-甲基-1-(1-甲基-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸甲酯(18ab)。根据上述一般方法,,将17a(100mg,0.62mmol)、2b(43mg,0.31mmol)和dipea(0.190ml,1.09mmol)在密封管中120℃加热6小时。在处理后,将己烷-etoac(85:15)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率66%(78mg);黄色固体;熔点=175至177℃;rf 0.40(2:1己烷-etoac);ir(kbr)3058(=c-h),2988-2851(c-h),1735(c=o),1704-1536(c=c),1409-1375(c-h弯曲),780(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl3)δ8.27(s,1h),7.89(d,j=8.0hz,1h),7.73(d,j=7.8hz,1h),7.56(dd,j=7.8,7.3hz,1h),7.50(d,j=8.0hz,1h),7.46(d,j=8.0hz,1h),7.33(dd,j=7.8,7.8hz,1h),7.19(m,2h),7.01(s,1h),4.53(q,j=7.0hz,2h),3.99(s,3h),3.79(s,3h),1.48(t,j=7.0hz,3h);
13
c nmr(100mhz,cdcl
3
)δ166.3,146.3,145.9,143.2,142.2,138.3,137.6,128.0,127.8,
123.1,122.4,121.2,121.1,120.9,120.8,119.8,109.8,109.1,105.5,104.4,61.9,31.1,29.5,14.5;hrms(esi)[c
24
h
21
n
3
o
2
+h
+
]计算值384.1707,实测值384.1672。
[0106]
示例21:5-甲基-1-(1-甲基-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸叔丁酯(18ac)。根据上述一般方法,,将17a(100mg,0.62mmol)、2c(52mg,0.31mmol)和dipea(0.190ml,1.09mmol)在密封管中120℃加热8小时。在处理后,将己烷-etoac(90:10)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗反应混合物;产率67%(85mg);黄色固体;熔点=200至202℃;rf0.60(1:1己烷-etoac);ir(kbr)3053(=c-h),2972-2852(c-h),1729(c=o),1686-1532(c=c),3.921412(c-h弯曲),781(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.14(s,1h),8.08(d,j=3.0hz,1h),7.73(d,j=3.0hz,1h),7.61-7.41(m,1h),7.37-7.28(m,1h),1.697.23(dd,j=7.06,2.4hz,1h),3.97(s,3h),3.89(m,3h),-3.60(m,9h);
13
c nmr(100mhz,cdcl3)δ165.2,146.2,146.1,144.3,142.2,138.4,137.7,127.8,120.9*,127.7,123.1,122.4,121.2,120.9,119.7,109.8,109.0,104.7,29.5;hrms(esi)[c
26
h
25
n
3
o
2
+h+]计算值325.1659,实测值412.2012。
*
高强度碳
[0107]
示例22:5-苄基-1-(1-苄基-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸甲酯(18ba)。根据上述一般方法,,将17b(0.100g,0.42mmol)、2a(26mg,0.21mmol)和dipea(0.095ml,0.74mmol)在密封管中120℃加热6小时。在处理后,将己烷-etoac(85:15)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率58%(63mg);黄色固体;熔点=168至170℃;rf 0.60(2:1己烷-etoac);ir(atr)3062(=c-h),2920-2850(c-h),1710(c=o),1667-1528(c=c),1467-1315(c-h弯曲),787-694(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.20(s,1h),8.13(d,j=8.0hz,1h),7.75(d,j=7.5hz,1h),7.48(dd,j=7.5,7.3hz,1h),7.41(m,2h),7.35-7.23(m,4h),7.20(d,j=7.5hz,1h),7.16(d,j=8.0hz,1h),7.14-7.08(m,3h),6.99-6.89(m,5h),5.67(s,2h),5.60(s,2h),3.98(s,3h);
13
c nmr(100mhz,cdcl
3
)δ166.8,146.5,145.8,142.9,141.7,138.3,138.1,136.9,135.6,129.0,128.1*,128.0,127.9,126.7,126.6,126.3,123.3,122.7,121.3,121.13,121.06,121.0,120.0,110.6,109.6,105.7,105.5,52.9,47.7,46.8;hrms(esi)c
35
h
27
n
3
o
2
+h
+
]计算值522.2176,实测值522.2160。
*
高强度碳
[0108]
示例23:5-苄基-1-(1-苄基-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸乙酯(18bb)。根据上述一般方法,,将17b(0.100g,0.62mmol)、2b(43mg,0.31mmol)和dipea(0.190ml,1.09mmol)在密封管中于120℃加热6小时。在处理后,将己烷-etoac(80:20)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率66%(78mg);红黄色液体;rf 0.40(2:1己烷-etoac);ir(kbr)3059(=c-h),2965-2860(c-h),1722(c=o),1609-1574(c=c),1423-1383(c-h弯曲),799(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.21-8.14(m,2h),7.75(d,j=7.5hz,1h),7.47(dd,j=7.8,7.3hz,1h),7.40(m,2h),7.35-7.23(m,4h),7.20(d,j=7.3hz,1h),7.16(d,j=7.3hz,1h),7.14-7.08(m,3h),7.02-6.89(m,5h),5.72(s,2h),5.59(s,2h),4.45(q,j=7.1hz,2h),1.40(t,j=7.1hz,3h);
13
c nmr(100mhz,cdcl
3
)δ166.2,146.5,145.9,143.2,141.7,138.4,138.1,136.9,135.7,129.0,128.2,128.1,128.0,127.8,126.7,126.6,126.4,123.3,122.7,121.2,121.1,121.0,120.9,120.0,110.6,109.6,105.52,105.48,61.8,47.7,46.8,14.4;hrms(esi)[c
36
h
29
n
3
o
2
+h
+
]计算值536.2333,实测值536.2349。
[0109]
示例24:5-苄基-1-(1-苄基-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸叔丁酯(18bc)。根据上述一般方法,,将17b(0.100g,0.62mmol)、2c(52mg,0.31mmol)和dipea(0.190ml,1.09mmol)在密封管中120℃加热8小时。在处理后,将己烷-etoac(85:15)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率66%(84mg);黄色固体;熔点=148至150℃;rf 0.60(2:1己烷-etoac);ir(atr)3062(=c-h),2926-2848(c-h),1706(c=o),1665-1531(c=c),1495-1323(c-h弯曲),782-694(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.28(d,j=8.0hz,1h),8.06(s,1h),7.75(d,j=7.8hz,1h),7.47(dd,j=7.5,7.3hz,1h),7.43-7.36(m,2h),7.34-7.22(m,4h),7.21-7.15(m,2h),7.15-7.10(m,3h),7.02-6.93(m,5h),5.86(s,2h),5.58(s,2h),1.63(s,9h);
13
c nmr(100mhz,cdcl
3
)δ165.2,146.4,146.1,144.5,141.7,138.6,138.1,137.0,135.8,129.0,128.2,128.0,127.84,127.75,126.7,126.5,126.4,123.3,122.7,121.2,121.1,120.9,120.4,120.0,110.6,109.6,105.7,104.9,81.8,47.5,46.8,28.2;hrms(esi)[c
38
h
33
n
3
o
2
+h
+
]计算值564.2646,实测值564.2644。
[0110]
示例25:8-甲氧基-1-(5-甲氧基-1-甲基-1h-吲哚-2-基)-5-甲基-5h-吡啶[4,3-b]吲哚-3-羧酸甲酯(18ca)。根据上述一般方法,,将17c(70mg,0.37mmol)、2a(23mg,0.18mmol)和dipea(0.110ml,0.63mmol)在密封管中120℃加热3小时。在处理后,将己烷-etoac(85:15)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率66%(84mg);黄色固体;熔点=160至162℃;rf 0.60(2:1己烷-etoac);ir(atr)3070(=c-h),2957-2850(c-h),1701(c=o),1660-1528(c=c),1485-1329(c-h弯曲),1105-991(c-o),810-688(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.26(s,1h),7.40(d,j=9.8hz,1h),7.32(d,j=8.8hz,1h),7.21-7.16(m,2h),7.15(d,j=2.0hz,1h),6.97(dd,j=9.0hz,2.3hz,1h),6.91(s,1h),4.05(s,3h),3.96(s,3h),3.89(s,3h),3.71(s,3h),3.55(s,3h);
13
c nmr(100mhz,cdcl
3
)δ167.0,154.8,154.3,146.3,145.8,142.6,137.9,137.2,133.8,128.1,121.2,120.8,117.7,112.8,110.4,109.9,105.8,105.0,103.8,102.5,55.8,55.7,53.0,31.1,29.6;hrms(esi)[c
25
h
23
n
3
o
4
+h
+
]计算值430.1761,实测值430.1764。
[0111]
示例26:5-(4-甲氧基苄基)-1-(1-(4-甲氧基苄基)-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸甲酯(18da)。根据上述一般方法,,将17d(0.100g,0.38mmol)、2a(24mg,0.19mmol)和dipea(0.120ml,0.66mmol)在密封管中120℃加热3小时。在处理后,将己烷-etoac(85:15)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率66%(84mg);黄色固体;熔点=106至108℃;rf 0.60(2:1己烷-etoac);ir(atr)3056(=c-h),2952-2835(c-h),1737(c=o),1664-1512(c=c),1457-1348(c-h弯曲),1106-989(c-o),819-695(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.22(s,1h),8.04(d,j=8.0hz,1h),7.73(d,j=7.8hz,1h),7.51-7.39(m,3h),7.27(d,j=7.3hz,1h),7.18(dd,j=7.5,7.0hz,1h),7.12(dd,j=7.3,7.3hz,1h),7.07(d,j=8.5hz,2h),7.06(s,1h),6.85(d,j=8.8hz,2h),6.83(d,j=8.8hz,2h),6.45(d,j=8.5hz,2h),5.55(s,2h),5.54(s,2h),3.99(s,3h),3.76(s,3h),3.52(s,3h);
13
c nmr(100mhz,cdcl
3
)δ166.9,159.4,158.4,146.6,145.7,142.9,141.7,138.0,137.0,130.4,128.1,127.95,127.92,127.75,127.68,123.3,122.6,121.2,121.1,121.04,121.01,119.9,114.5,113.5,110.6,109.6,105.7,105.3,55.3,55.1,52.9,47.2,46.4;hrms(esi)[c
37
h
31
n
3
o
4
+h
+
]计算值582.2387,实测值582.2373。
[0112]
示例27:5-丁基-1-(1-丁基-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸甲酯(18ea)。根据上述一般方法,,将17e(0.100g,0.49mmol)、2a(31mg,0.25mmol)和dipea(0.114ml,0.88mmol)在密封管中于120℃加热3小时。在处理后,将己烷-etoac(85:15)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率66%(84mg);黄色液体;rf 0.60(2:1己烷-etoac);ir(kbr)3064(=c-h),2972-2854(c-h),1726(c=o),1621-1570(c=c),1462-1317(c-h弯曲),796(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.27(s,1h),8.00(d,j=8.0hz,1h),7.72(d,j=7.5hz,1h),7.59-7.45(m,3h),7.30(dd,j=7.6,7.5hz,1h),7.18(dd,j=8.2,7.2hz,1h),7.13(dd,j=8.2,7.3hz,1h),6.97(s,1h),4.43(t,j=6.8hz,2h),4.34(t,j=7.0hz,2h),4.05(s,3h),2.00-1.87(m,2h),1.71-1.60(m,2h),1.52-1.39(m,2h),1.16-1.04(m,2h),0.99(t,j=7.0hz,3h),0.62(t,j=7.3hz,3h);
13
c nmr(100mhz,cdcl
3
)δ167.1,146.8,145.4,142.7,141.6,137.7,136.9,127.92,127.87,123.3,122.2,121.3,120.9,120.8*,119.6,110.2,109.3,105.6,104.7,52.9,43.9,43.4,32.2,31.1,20.6,20.0,13.9,13.5;hrms(esi)[c
29
h
31
n
3
o
2
+h
+
]计算值454.2489,实测值454.2559。
*
合并的碳。
[0113]
示例28:5-丁基-1-(1-丁基-1h-吲哚-2-基)-5h-吡啶[4,3-b]吲哚-3-羧酸甲酯(18fa)。根据上述一般方法,,将17f(0.100g,0.33mmol)、2a(21mg,0.17mmol)和dipea(0.101ml,0.58mmol)在密封管中120℃加热8小时。在处理后,将己烷-etoac(90:10)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率63%(70mg);黄色固体;熔点=205至207℃;rf 0.60(2:1己烷-etoac);ir(atr)3413(n-h),3062(=c-h),2956-2850(c-h),1706(c=o),1633-1489(c=c),1448-1350(c-h弯曲),1307(n-s=o),1145(s=o),812-687(=c-h bend)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.16(dd,j=8.0,8.0hz,2h),7.73(d,j=8.5hz,2h),7.70(d,j=8.5hz,2h),7.32-7.26(m,3h),7.24-7.19(m,3h),7.18-7.14(m,2h),7.09(d,j=2.0hz,1h),6.98(dd,j=7.5,7.3hz,1h),6.59(d,j=2.0hz,1h),6.38(d,j=7.8hz,1h),5.90(s,1h),5.64(s,1h),3.83(s,3h),2.41(s,3h),2.37(s,3h);
13
c nmr(100mhz,cdcl3)δ163.9,145.4,145.0,139.4,137.7,137.4,136.3,135.2,135.0,132.6,130.1,129.9,128.8,127.7,126.8,126.3,125.2,124.4,124.0,123.8,121.2,117.5,114.9,114.8,113.1,110.6,94.4,52.6,47.9,21.7,21.6;hrms*(esi)[c
35
h
27
n
3
o
6
s
2
+h
+
]计算值650.1414,实测值650.1386。
*
hrms峰对应于化合物18fa的脱氢或芳构化形式。
[0114]
示例29:4-(呋喃-2-基)呋喃并[3,2-c]吡啶-6-羧酸甲酯(20aa)。根据上述一般方法,,将19a(200mg,2.08mmol)、2a(130mg,1.04mmol)和dipea(0.670ml,3.72mmol)在密封管中120℃加热6小时。在处理后,将己烷-etoac(98:2)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率50%(126mg);灰白色固体;熔点=95至97℃;rf 0.65(4:1己烷-etoac);ir(kbr)3032(=c-h),2957-2856(c-h),1731(c=o),1713-1560(c=c),1359(c-h弯曲),1112-993(c-o),722(=c-h弯曲)cm-1;1h nmr(400mhz,cdcl
3
)δ8.19(s,1h),7.85(d,j=3.0hz,1h),7.65(dd,j=1.9,0.92hz,1h),7.45-7.40(m,1h),7.36(d,j=3.0hz,1h),6.61(dd,j=3.5hz,1.9hz,1h),4.03(s,3h);
13
c nmr(100mhz,cdcl
3
)δ166.0,160.6,153.2,148.1,144.2,143.5,143.2,122.7,112.2,111.4,107.9,106.9,53.0;hrms(esi)[c
13
h
9
no
4
+na
+
]计算值266.0424的,实测值266.0417
[0115]
示例30:4-(呋喃-2-基)呋喃并[3,2-c]吡啶-6-羧酸乙酯(20ab)。根据上述一般方
法,,将呋喃-2-甲醛(19a,200mg,2.08mmol)、甘氨酸乙酯(2b,145mg,1.04mmol)和dipea(0.670ml,3.72mmol)在密封管中于120℃加热6小时。在处理后,将己烷-etoac(99:1)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率48%(128mg);黄色液体;rf 0.70(4:1己烷-etoac);ir(kbr)3032(=c-h),2976-2855(c-h),1742(c=o),1730-1524(c=c),1371(c-h弯曲),1165-1005(c-o),741(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.17(s,1h),7.83(d,j=2.0hz,1h),7.64(m,1h),7.42(dd,j=2.0hz,0.8hz,1h),7.37(d,j=3.2hz,1h),6.60(dd,j=3.3hz,1.8hz,1h),4.49(q,j=7.3hz,2h),1.47(t,j=7.3hz,3h);
13
c nmr(400mhz,cdcl3)δ165.4,160.7,153.4,148.1,144.1,143.6,143.5,122.5,112.3,111.4,107.8,106.9,62.0,14.4;hrms(esi)[c
14
h
11
no
4
+na
+
]计算值280.0580,实测值280.0571。
[0116]
示例31:4-(呋喃-2-基)呋喃并[3,2-c]吡啶-6-羧酸叔丁酯(20ac)。根据上述一般方法,,将19a(200mg,2.08mmol)、2c(175mg,1.04mmol)和dipea(0.670ml,3.72mmol)在密封管中120℃加热7小时。在处理后,将己烷-etoac(99:1)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率46%(136mg);灰白色固体;熔点=78至80℃;rf 0.70(4:1己烷-etoac);ir(kbr)3032(=c-h),2976-2855(c-h),1742(c=o),1730-1524(c=c),1371(c-h弯曲),1165-1005(c-o),741(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.08(s,1h),7.82(d,j=3.0hz,1h),7.63(dd,j=1.5,0.5hz,1h),7.43(dd,j=2.0hz,0.8hz,1h),7.39(d,j=3.0hz,1h),6.60(dd,j=3.5hz,1.8hz,1h),1.66(s,9h);
13
c nmr(400mhz,cdcl3)δ164.0,160.7,153.7,147.9,144.6,143.9,143.4,122.2,112.2,111.2,107.4,106.9,82.0,28.2;hrms(esi)[c
16
h
15
no
4
+na
+
]计算值308.0893,实测值308.0890。
[0117]
示例32:2-甲基-4-(5-甲基呋喃-2-基)呋喃并[3,2-c]吡啶-6-羧酸甲酯(20ba)。根据上述一般方法,,将5-甲基呋喃-2-乙醛(19b,200mg,1.82mmol)、甘氨酸甲酯盐酸盐(2a,114mg,0.91mmol)和dipea(0.555ml,3.18mmol)在密封管中120℃加热6小时。在处理后,将己烷-etoac(99:1)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率54%(133mg);灰白色固体;熔点=78至80℃;rf 0.70(4:1己烷-etoac);ir(kbr)3032(=c-h),2976-2855(c-h),1742(c=o),1730-1524(c=c),1371(c-h弯曲),1165-1005(c-o),741(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.04(s,1h),7.17(d,j=3.0hz,1h),6.96(s,1h),6.18(d,j=3.0hz,1h),4.00(s,3h),2.55(s,3h),2.45(s,3h);13c nmr(400mhz,cdcl
3
)δ166.2,160.3,159.4,154.2,151.5,142.3,142.2,124.0,112.3,108.5,106.8,102.9,52.8,14.2,14.0。
[0118]
示例33:(2-甲基-4-(5-甲基呋喃-2-基)呋喃并[3,2-c]吡啶-6-基)甲醇(21)。在双颈圆底烧瓶(50ml)中,在惰性气氛下将2-甲基-4-(5-甲基呋喃-2-基)呋喃并[3,2-c]吡啶-6-羧酸甲酯(20ba,0250gms,0,92mmol)溶解在干燥thf(5ml)中。将反应混合物冷却至0℃,并小心地将单份固体lialh4(0.104gms,2.76mmol)添加到其中。将反应混合物加热至室温并搅拌20分钟。通过tlc监控反应过程。再次将反应混合物冷却至0℃,通过添加饱和nh
4
cl(10ml)溶液来淬火多余的lialh
4
。用etoac(10
×
3ml)提取水层,并在无水na
2
so
4
上干燥合并的有机提取物。过滤粗品,并在减压条件下蒸发溶剂。以己烷-etoac(80:20)为洗脱液,采用中性矾土(175目)柱层析法,纯化残留物;产率66%(148mg);黄色液体;rf0.45(1:1正己烷-etoac);
1
h nmr(400mhz,cdcl3)δ7.15-7.05(m,2h),6.90(s,1h),6.17(d,j=
1.7hz,1h),4.80(s,2h),2.50(s,3h),2.45(s,3h);
13
c nmr(400mhz,cdcl
3
)δ161.2,156.7,153.8,153.2,152.3,140.9,120.4,111.1,108.3,102.3,100.9,64.3,14.04,14.01。
[0119]
示例34:2-甲基-4-(5-甲基呋喃-2-基)-6-((丙炔-2-炔-1-氧基)甲基)呋喃[3,2-c]吡啶(22)。在双颈圆底烧瓶(50ml)中,在惰性气氛下将(2-甲基-4-(5-甲基呋喃-2-基)呋喃并[3,2-c]吡啶-6-基)甲醇(21,70mg,0,29mmol)溶解在干燥dmf(2ml)中。将反应混合物冷却至0℃,并将单份氢化钠(55-60%悬浮在矿物油中,14mg,0.35mmol)添加到其中。将反应混合物在相同温度下搅拌20分钟。通过微量移液管逐滴添加丙炔溴(80%溶于甲苯,33μl,0.35mmol)。将反应混合物加热至室温并进一步搅拌3小时。通过tlc监控反应过程。将反应混合物短暂冷却,然后通过添加盐水(5ml)淬火反应。用etoac(10
×
3ml)提取水层,并将合并的有机提取物在无水na
2
so
4
上干燥。过滤粗品,并在减压条件下蒸发溶剂。采用己烷-etoac(97:3)用作洗脱液来采用中性矾土(175目)柱层析法,纯化残留物;产率70%(57mg);黄色油状液体;rf 0.70(7:3己烷-etoac)。
[0120]
示例35:1-(2-(4-((2-甲基-4-(5-甲基呋喃-2-基)呋喃并[3,2-c]吡啶-6-基)甲氧基)甲基)-1h-1,2,3-三唑-1-基)乙基)吲哚-2,3-二酮(26)。在圆底瓶(25ml)中,将2-甲基-4-(5-甲基呋喃-2-基)-6-((丙炔-2-炔-1-氧基)甲基)呋喃并[3,2-c]吡啶(22,30mg,0.11mmol)和1-(2-叠氮乙基)吲哚-2,3-二酮(25,24mg,0.11mmol)溶解于dmf(2ml)中。在惰性气氛下,在室温下将一份乙酸铜(ii)(一水化,12mg,0.06mmol)加入上述溶液中。将反应混合物在室温下继续搅拌24小时。通过tlc监测反应过程。加入盐水(5ml)淬灭反应,用etoac(5
×
3ml)萃取水层。将合并的有机提取物在无水na
2
so
4
中干燥。过滤粗品,并在减压条件下蒸发多余溶剂。以ch
2
cl
2-meoh(95:5)溶剂混合物用作洗脱液来采用中性矾土(175目)柱层析法,纯化残渣;产率82%(45mg),黄橙色液体;rf0.25(1:3正己烷-etoac);
1
h nmr(400mhz,cdcl
3
)δ7.62(s,1h),7.51(d,j=7.5hz,1h),7.45(dd,j=7.8,7.5hz,1h),7.25(s,1h),7.05-6.95(m,2h),6.87(s,1h),6.52(d,j=8.0hz,1h),6.17(d,j=2.8hz,1h),4.75-4.68(m,4h),4.67(s,2h),4.25(t,j=5.8hz,2h),2.52(s,3h),2.45(s,3h);hrms(esi)[c
27
h
23
n
5
o
5
+na+]计算值520.1591,实测值520.1595。
[0121]
示例36:1-(苯并呋喃-2-基)苯并呋喃并[3,2-c]吡啶-3-羧酸甲酯(28aa)。根据上述一般方法,,将27(100mg,0.68mmol)、2a(43mg,0.34mmol)和dipea(0.240ml,1.36mmol)在密封管中120℃加热6小时。在处理后,将己烷-etoac(98:2)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率60%(70mg);黄橙色固体;熔点=163至165℃;rf 0.40(8:2己烷-etoac);ir(kbr)3065(=c-h),2948-2850(c-h),1720(c=o),1612-1539(c=c),1350-1338(c-h弯曲),1256-1094(c-o),735(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.85(d,j=7.8hz,1h),8.30(s,1h),7.81(s,1h),7.78-7.71(m,2h),7.69(d,j=8.0hz,1h),7.63(dd,j=8.0,7.3hz,1h),7.51(dd,j=7.6,7.5hz,1h),7.44(dd,j=7.8,7.5hz,1h),7.34(dd,j=7.6,7.5hz,1h),4.08(s,3h);
13
c nmr(400mhz,cdcl
3
)δ165.5,162.7,157.4,155.4,154.5,145.4,144.3,129.6,128.4,125.7,125.6,124.3,123.7,122.1,120.9,120.8,111.9,111.5,108.7,108.4,53.1;hrms(esi)[c
21
h
13
no
4
+na
+
]计算值366.0737,实测值366.0693。
[0122]
示例37:1-(苯并呋喃-2-基)苯并呋喃并[3,2-c]吡啶-3-羧酸乙酯(28ab)。根据上述一般方法,,将27(100mg,0.68mmol)、2b(48mg,0.34mmol)和dipea(0.240ml,1.36mmol)在
密封管中120℃加热6小时。在处理后,将己烷-etoac(98:2)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率55%(67mg);黄色固体;熔点=141至143℃;rf 0.40(8:2己烷-etoac);ir(kbr)3065(=c-h),2987-2850(c-h),1714(c=o),1625-1540(c=c),1367-1340(c-h弯曲),1266-1097(c-o),750(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl3)δ8.85(d,j=8.0hz,1h),8.26(s,1h),7.81(s,1h),7.75(d,j=8.0hz,1h),7.72(d,j=7.8hz,1h),7.67(d,j=8.3hz,1h),7.61(dd,j=8.0,7.5hz,1h),7.49(dd,j=8.0,8.3hz,1h),7.43(dd,j=7.8,7.5hz,1h),7.34(dd,j=8.0,8.3hz,1h),4.54(q,j=7.1hz,2h),1.51(t,j=7.1hz,3h);
13
c nmr(100mhz,cdcl3)δ164.9,162.8,157.4,155.5,154.7,145.8,144.3,129.6,128.5,125.74,125.73,124.3,123.7,122.2,121.0,120.7,111.9,111.6,108.7,108.3,62.2,14.4;hrms(esi)[c
22
h
15
no
4
+na+]计算值380.0893,实测值380.0844。
[0123]
示例38:1-(苯并呋喃-2-基)苯并呋喃[3,2-c]吡啶-3-羧酸叔丁酯(28ac)。根据上述一般方法,,将27(100mg,0.68mmol)、2c(57mg,0.34mmol)和dipea(0.240ml,1.36mmol)在密封管中120℃加热7小时。在处理后,将己烷-etoac(98:2)用作洗脱液来通过矾土(中性,175目)柱层析法纯化粗品;产率45%(64mg);黄色固体;熔点=100至102℃;rf 0.40(8:2己烷-etoac);ir(kbr)3060(=c-h),2977-2851(c-h),1715(c=o),1626-1540(c=c),1365-1340(c-h弯曲),1273-1074(c-o),738(=c-h弯曲)cm-1;
1
h nmr(400mhz,cdcl
3
)δ8.88(d,j=8.0hz,1h),8.18(s,1h),7.83(s,1h),7.747.71(m,1h),7.65(d,j=8.0hz,1h),7.59(dd,j=8.0,7.3hz,1h),7.48(dd,j=7.6,7.5hz,1h),7.42(dd,j=8.3,7.5hz,1h),7.33(dd,j=7.6,7.5hz,1h),1.71(s,9h);
13
c nmr(400mhz,cdcl
3
)δ163.6,163.6,162.8,157.4,155.4,154.9,146.8,144.1,129.3,128.5,125.7,125.6,124.2,123.6,122.1,121.0,120.2,111.8,111.5,108.6,107.9,82.4;hrms(esi)[c
24
h
13
no
4
+na
+
]计算值366.0737,实测值408.1153。
[0124]
上述对具体实施例的描述将充分揭示本文中实施例的一般性质,使得其他人可以通过应用现有知识,在不脱离一般概念的前提下,容易地修改和/或适应这些特定实施例的各种应用,因此,此类适应和修改应当以及旨在在所公开实施例的等效含义和范围内理解。应当理解的是,本文中使用的措辞或术语是为了描述而非限制目的。因此,虽然已经根据优选实施例描述了本文实施例,但是本领域技术人员将认识到,可以在如本文的实施例的主旨和范围内进行修改来实践本文中的实施例。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips