
 商标分类
商标分类  商标转让
商标转让 
含独特分子标签序列的转座酶复合体及其应用的制作方法
 2021-02-02 13:02:07|
2021-02-02 13:02:07| 644|
644| 起点商标网
起点商标网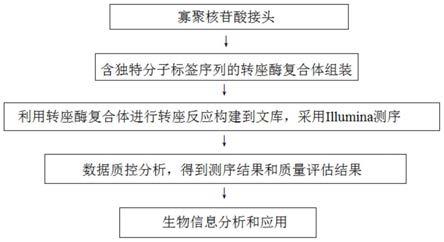
[0001]
本发明涉及测序技术领域,具体涉及一种含独特分子标签序列(umi)的转座酶复合体及其应用。
背景技术:
[0002]
新一代测序(next generation sequencing,ngs)技术深刻影响了生物学研究和临床诊断。构建测序文库是进行ngs的基础。从2010年开始,转座酶tn5被广泛用于构建基于illumina公司的测序平台的dna测序文库。其原理是:tn5是一种细菌转座子,最早在大肠杆菌中发现。经改造后,tn5转座酶可以与长度为19bp的me(mosaic end)双链序列结合,并将其近乎随机地插入双链dna大片段中。因此,只要将接头与me序列一起合成,然后与tn5转座酶孵育,形成转座复合体,再在适宜的体系和条件下与dna样本孵育,就可一步实现dna样本片段化和接头连接,极大地减少了所需样本量,提高了建库的效率。
[0003]
2013年,jason d buenrostro等提出atac-seq(assay for transposase-accessible chromatin using sequencing)方法,将转座酶tn5与真核生物的细胞核一起孵育,通过检测tn5插入基因组各区段的频率,可以评估染色质开放程度(即“染色质可及性”)。该方法具有快速、灵敏的特点,已被广泛用于表观基因组研究。在atac-seq结果中,tn5插入事件的密度反映了一个区域的染色质的可及性;而在一个高度可及的区域中,一些插入事件突然下降的小区域可能表征了结合有转录因子,称为“足迹(footprint)”。
[0004]
2020年,lin di等和bo lu等相继发现转座酶tn5可插入rna/dna杂合双链,提出其可用于快速构建rna测序文库。
[0005]
与大多数ngs建库测序的方法一样,上述基于tn5转座酶的应用,均需要使用pcr扩增,以确保获得足够的dna片段在illumina平台上测序。由于不同片段的扩增效率不同,在最终的测序文库中可能某些片段的数量会过多,为后续的一些分析带来偏差。为了消除这种偏差,我们通常将与基因组比对后具有相同的基因组坐标的读段(read)认为是pcr引起的重复拷贝,并在各种分析之前将其去除。然而,这种重复数据删除模式在某些情况下是有缺陷的。例如,随着测序深度的增加,不同的tn5插入事件产生相同片段的概率会增加。对于小基因组或具有许多串联重复的物种,这种现象会加剧,因为它们在pcr扩增之前就产生相同片段的概率要高得多。在这种情况下,基于基因组坐标删除pcr重复将会误删有信息的片段,也会引入偏差。
[0006]
umi(unique molecular identifiers)是一种分子标签,由随机合成的序列组成,常用来对mrna转录本进行准确定量。在单细胞rna-seq中,mrna文库通过逆转录过程,在5
’
端加入带有umi的引物标签,再进行pcr扩增和测序。测序结果中不同的mrna转录本将带有不同的umi标签,而通过pcr产生的拷贝,将带有相同的umi标签。因此,在生物信息分析过程中,umi作为内部验证对照,可以有效去除由pcr引起的重复拷贝,从而做到准确定量。目前,尚未有将umi技术与tn5转座酶建库技术结合的报道。
[0007]
最后,构建好测序文库后,上机测序时,需要选择合适的、能跟文库片段匹配的测
序引物。目前illumina测序平台的通用测序引物有两种类型,分别叫truseq和nextera。其中基于转座酶tn5构建的文库,使用的测序引物是nextera,其主要序列是转座酶结合序列me,其原因是me序列在tn5转座时插入dna样本,其3
′
与dna样本相连。因此,测序时将从me序列3
′
开始读取信号。为了不影响转座反应,只能将umi与me的5
′
相连,此时用nextera测序引物将无法获得umi序列。另一方面,只有样本建库时包含与通用测序引物匹配的序列,才能方便与不同研究者的测序文库混合,一起进行商业化测序,降低成本、提高效率;否则,需要使用自定义测序引物,承包流动槽(lane)测序,存在灵活性和成本的问题。因此,精巧地设计tn5转座酶复合体的接头序列,是实现umi技术与tn5转座酶建库技术结合并商业化推广应用的关键。
[0008]
专利cn103938277b和cn110628889a公开了两种含随机碱基(即umi)的测序文库的构建方法,但其读取随机碱基的方式是通过索引引物(index primer)进行的,占据了区分不同样品的索引序列,给商业化测序带来限制。
技术实现要素:
[0009]
本发明第一目的是提供了一种含独特分子标签序列(umi)的转座酶复合体,该转座酶复合体具有以下特点:
[0010]
1)umi不占用样品索引序列,便于使用商业化测序服务;
[0011]
2)读取umi的方式不同(通过read1primer或read2 primer读取),从而允许更长的umi。
[0012]
为实现上述目的,本发明所设计一种含独特分子标签序列的转座酶复合体,所述转座酶复合体为由寡聚核苷酸接头和转座酶组装形成复合体;其中,
[0013]
寡聚核苷酸接头的第一链内含有一个umi标签序列,
[0014]
或,含有多个依次连接的umi标签序列组成umi复合标签序列且umi标签序列为一种或多种;
[0015]
或,含有多个由间隔序列分隔的umi标签序列组成umi复合标签序列且umi标签序列为一种或多种;其中,间隔序列由长度为0~20bp的可替换的非随机的序列组成,gc含量为25%-75%。
[0016]
进一步地,所述寡聚核苷酸接头的第一链从5
′
端到3
′
端依次包括通用接头序列、间隔序列、umi标签序列/umi复合标签序列、间隔序列、条码序列和me序列;间隔序列为0-20bp的序列;
[0017]
所述寡聚核苷酸接头的第二链为包含me序列的互补序列(第二链的长度不超过第一链的长度)。
[0018]
再进一步地,所述通用接头序列为10-50bp且序列(dna序列)中,gc含量为25%-75%(其作用为与之后建库所用pcr引物结合);
[0019]
umi标签序列为2-100bp的序列(用于区分转座酶插入重复事件和pcr重复),条码序列为0-100bp的序列(用于区分不同样品或保持碱基组成平衡),me序列用于与转座酶结合的序列(不同的酶选用不同的me序列),间隔序列为0-20bp的序列(用于间隔通用接头、多个umi及条码序列,以及用于区分不同样品或保持碱基组成平衡)。
[0020]
再进一步地,所述通用接头序列为含有“ttccgatct”的序列。
[0021]
再进一步地,所述寡聚核苷酸接头的第一链的表达式为:
[0022]
[]ttccgatct{}<>agatgtgtataagagaca,
[0023]
其中,[]ttccgatct为通用接头序列,其长度为10~50个核苷酸;
[0024]
{}为umi标签序列或umi复合标签序列,
[0025]
若{}为umi标签序列,其长度为2~50个核苷酸;
[0026]
或,若{}为umi复合标签序列,其长度为4~50个核苷酸;
[0027]
<>为条码序列,其长度为0~50个核苷酸;
[0028]
agatgtgtataagagaca为me序列;
[0029]
再进一步地,所述寡聚核苷酸接头的第一链选自下列序列中任意一条:
[0030]
[0031][0032]
其中,cgacgctcttccgatct为通用接头序列,agatgtgtataagagacag为me序列;
[0033]
含有“n”的序列均为{}<>的组合。
[0034]
作为最优选方案,所述寡聚核苷酸接头的第一链选自下列序列中任意一条:
[0035]
me-a6cgacgctcttccgatctnnnnnnagatgtgtataagagacagme-a13cgacgctcttccgatctnnnnnnnnnnnnnagatgtgtataagagacagme-a20cgacgctcttccgatctnnnnnnnnnnnnnnnnnnnnagatgtgtataagagacagme-a25cgacgctcttccgatctnnnnnnnnnnnnnnnnnnnnctgctagatgtgtataagagacag
[0036]
再进一步地,所述转座酶(商品化或自主表达的酶)选自tn5转座酶、tn5变体、mu、mu e392q、vibhar、rag和tn552。
[0037]
本发明第二目的是提供了上述转座酶复合体在rna测序文库构建、dna测序文库构建和染色质可及性测序(atac-seq)文库的构建中的应用。
[0038]
本发明提供了一种用于构建测序文库的试剂盒,它包括复合体组合物、转座反应缓冲液、pcr引物、pcr酶和pcr扩增缓冲液;其中,所述复合体组合物包含一种上述的转座酶复合体/多种上述的转座酶复合体。
[0039]
利用上述试剂盒构建含umi的染色质可及性测序(umi-atac-seq)文库的方法包括以下步骤:
[0040]
1)材料准备和提取细胞核;
[0041]
2)利用试剂盒转座、纯化和pcr,得到pcr产物;
[0042]
3)pcr产物测序:
[0043]
将pcr产物磁珠纯化后,采用illumina系列平台进行pe150测序;其中,pcr产物的序列结构如下:
[0044]
p5—index 5—universal connector a—umi—barcode—me—dna insert—me—universal connector b—index 7—p7;
[0045]
公式中,p5序列和p7序列是illumina标准文库要求的序列,与测序芯片(flowcell)上固定的序列互补或一致;
[0046]
index 5和index 7序列为6-8bp可替换的dna序列(用于多个文库混合测序时区分不同的文库,可以对多个样品建库后混池测序);
[0047]
测序时根据universal connector a和universal connector b来设计对应的read1和read2的测序引物,index 5和index 7的测序引物同illumina测序仪所用;
[0048]
3)对测序结果进行生物信息分析,得到数据质控结果。
[0049]
本发明的有益效果:
[0050]
1.利用本发明所述的含有umi标签的转座酶复合体构建的二代测序文库,能够有效区分转座酶插入重复事件和pcr导致的重复读段,提高了测序数据的利用率,提高测序读段的利用率、序列变异及其频率鉴定的可靠性、基因表达和染色质可及性定量的准确性,以及鉴定dna结合蛋白的足迹(footprint)的敏感性。
[0051]
2.利用本发明构建的含有umi标签的转座酶复合体,可以构建兼容主流测序仪的文库,便于商业化推广应用。
[0052]
3.本发明设计的umi标签长度为2-100bp。通过不同长度umi组合,有利于文库碱基平衡性,可以有效提高测序质量和兼容性。
[0053]
4.本发明设计的兼容标准truseq文库的接头,可将所构建的文库与标准illumina truseq或nextera文库一起混合上机测序,不需要使用自定义测序引物,可以降低成本、提高效率。
附图说明
[0054]
图1为含独特分子标签序列的转座酶复合体及其构建文库和应用的流程图;
[0055]
图2为含有umi和条码的寡核苷酸接头的第一链的结构示意图;
[0056]
图3为含有umi接头的illumina测序文库的结构示意图;
[0057]
图4为实施例用含umi的转座酶复合体进行的atac-seq实验的流程图;
[0058]
图中,图4a为组装含umi的转座酶复合体的示意图;
[0059]
图4b为atac-seq实验的流程图,流程步骤依次为获取细胞核、与转座酶复合体孵育、纯化dna、pcr构建文库和上机测序;
[0060]
图5为样本c019中具有相同比对位点但umi不同的被挽救读段的分布图;通常的测序文库中,比对位点相同的读段通常会被当成pcr重复而被去除,而如果使用含umi的转座酶复合体构建文库,根据其所含umi不同,部分读段可以被“挽救”;
[0061]
图中,框图说明了umi可以在高染色质可及性的基因组区段挽救更多的读段。将基
因组划分为1kb的区块,并计算每个区块中被挽救的读段的比例,然后将所有的区块按25%、50%和75%的量级进行分组,获得此图。本分析中只有比对质量大于三十的读段被使用;
[0062]
图6为不同方法去除pcr重复的定量结果相关性图;
[0063]
图中,通过散点图对比了无重复数据删除(nd)、基于坐标的重复数据删除(cd)和基于umi的重复数据删除(ud)数据集的量化结果;每个点是250bp区块(对数基数2)中tn5插入的数量;
[0064]
图7为umi的使用提高了转录因子足迹识别的灵敏度图;
[0065]
图中,图7a为显示了数据经cd和ud方式处理后,由pydnase确定的足迹(p值<10-30
)的维恩图,其中,共同的足迹被定义为至少有一个碱基重叠的足迹;
[0066]
图7b数据经cd和ud方式处理后,pydnase计算获得的足迹分数的关系图;其中,足迹分数的计算方式为log(-log
10
(p值)),点的颜色与a中的维恩图相同,小图显示的是仅cd方式处理后鉴定的足迹;
[0067]
图7c为数据经cd(蓝色)、ud(红色)或nd(灰色)方式处理后,一些鉴定的足迹周围的tn5插入频率;阴影区域对应的基因组坐标显示在每个图的上方;
[0068]
图7d为足迹深度(fpd)计算的示意图,其中,"avg"代表平均值函数;
[0069]
图7e为数据经cd和ud方式处理后,计算的fpd值的差异的分布图,所涉及的足迹是指数据经cd方式处理后鉴定的足迹。
具体实施方式
[0070]
下面结合具体实施例对本发明作进一步的详细描述,以便本领域技术人员理解。
[0071]
实施例1
[0072]
含独特分子标签序列的转座酶复合体构建(组装):
[0073]
1.设计寡聚核苷酸接头
[0074]
me-a6cgacgctcttccgatctnnnnnnagatgtgtataagagacagmerev[phos]ctgtctcttatacacatct
[0075]
注:me-a6为寡聚核苷酸接头的第一链,其中cgacgctcttccgatct为通用接头序列,nnnnnn为umi序列,agatgtgtataagagacag为me序列,与形成完整接头所需的第二链merev反向互补。
[0076]
2.转座酶复合体组装
[0077]
a.将merev和me-a6按照1:1混合,一次混合20μl;
[0078]
b.将混合的引物经过高温变性(95℃3min)后,逐渐降温退火至12℃,获得5
′
粘性末端的接头meds-a6,即为组装所用接头。
[0079]
c.按照如下比例,将上述合成的接头与tn5酶(tn5酶可通过商业化途径购买获得)混合:30μl tn5酶,4.2μl meds-a6(100μm);25℃,孵育60min,即得到转座酶复合体tn5-umi-a6,-20℃保存。
[0080]
按上述方法将寡聚核苷酸接头的第一链替换为下述表中的寡聚核苷酸,可组装成各种转座酶复合体:
[0081]
表1
[0082][0083][0084]
实施例2
[0085]
试剂盒包括实施例2获得的复合体组合物,缓冲液,pcr引物,pcr酶和pcr扩增缓冲液;
[0086]
转座反应缓冲液体系为50mm tris-hcl、25mm mgcl
2
和50%v/v dmf或其他适合tn5转座反应的缓冲液。
[0087]
pcr引物为:
[0088][0089]
其中下划线部分为可替换的条码序列。
[0090]
pcr酶为高保真酶,如high-fidelity 2x pcr master mix。
[0091]
上述试剂盒中的复合体组合物制备方法如下:
[0092]
1.设计寡聚核苷酸接头集合
[0093]
合成融合umi的tn5组装所需接头序列,见表2。
[0094]
表2.tn5接头序列引物
[0095][0096][0097]
注:me-a25中的小写字符碱基为调节碱基平衡的条码序列。me-b为文献(jason d buenrostro等,nature methods,2013,10:1213)所述序列,与merev形成另一种寡聚核苷酸接头,便于pcr构建测序文库。
[0098]
2.复合体组合物的组装(图4a)
[0099]
a.将上述引物按照如下组合1:1混合,meds-a6(merev+me-a6)、meds-a13(merev+me-a13)、meds-a20(merev+me-a20)、meds-a25(merev+me-a25)、meds-b(merev+me-b),每组引物对应一个离心管,分别混合20μl。
[0100]
b.将混合的引物经过高温变性(95℃3min)后,逐渐降温退火至12℃,获得组装所用5
′
粘性末端的接头;
[0101]
c.将上述四种接头meds-a6、meds-a13、meds-a20、meds-a25等摩尔混合为meds-a;
[0102]
d.按照如下比例,将合成的接头与tn5酶混合:30μl tn5,2.1μl meds-a(100μm),2.1μl meds-b(100μm)。25℃,孵育60min,得到复合体混合物,-20℃保存。
[0103]
实施例3
[0104]
以水稻叶片为材料,利用实施例2的试剂盒构建含有umi接头的atac-seq文库(以下称为umi-atac-seq):
[0105]
步骤s1,材料准备和文库构建(图4b)
[0106]
a.水稻叶片细胞核制备:取新鲜水稻叶片,加入500μl细胞裂解液,用刀片快速剁
碎,过滤后加入0.1%dapi染色。用流式细胞仪分选100,000个细胞核至1.5ml离心管中,离心收集细胞核,冰上备用。
[0107]
b.转座反应和纯化:在冰上按照如下比例配置反应体系:2μl复合体组合物,8μl 5x tagmentation buffer,30μl ddh
2
o。吸打混匀后,37℃,孵育30min。反应完成后用takara minibest dnafragment purification kit(no.9761)纯化。
[0108]
c.pcr扩增:采用high-fidelity 2x pcr master mix(m0541)进行,反应体系如下:纯化dna 20μl,2x pcr master mix25μl,pcr primer 1(10μm)2.5μl,barcoded pcr primer 2(10μm)2.5μl。pcr反应程序为:72℃3min,98℃30sec;98℃10sec,63℃30sec,72℃1min,11cycles;72℃5min。
[0109]
pcr所用引物序列见表3。
[0110]
表3.pcr扩增引物
[0111][0112]
注:[i7]为标签序列,长度为8bp,本例中使用的标签序列共有7个,为tgcctctt、tcctctac、atcacgac、acagtggt、cagatcca、acaaacgg、acccagca。用于针对多样品混合测序时区分不同样品的测序读段。
[0113]
d.pcr产物测序:将pcr产物磁珠纯化后,采用illumina hiseq x-ten平台进行pe150测序;其中,pcr产物的序列结构如下:
[0114]
p5—index 5—universal connector a—umi—barcode—me—dna insert—me—universal connector b—index 7—p7(图3)。
[0115]
上述结构中,p5序列、p7序列是illumina标准文库的要求,与测序芯片(flowcell)上固定的序列互补或一致。
[0116]
index 5和index 7序列为6-8bp dna固定序列,用于多个文库混合测序时区分不同的文库,可以对多个样品建库后混池测序。
[0117]
测序时根据universal connector a和universal connector b来设计对应的read1和read2的测序引物,index 5和index 7的测序引物同illumina测序仪所用。
[0118]
本实施例中,read1测序引物为
[0119]“acactctttccctacacgacgctcttccgatct”,即illumina测序试剂盒中包含的truseq标准测序引物。
[0120]
步骤s2,对测序结果进行生物信息分析,得到数据质控结果。用fastqc对测序数据进行质量统计;去除测序接头后,利用bwa将测序数据比对至水稻参考基因组,统计比对率;samtools去除重复及低质量比对的序列并统计重复率。
[0121]
步骤s3,评估umi的应用对数据利用率的影响
[0122]
表4 umi-atac-seq测序数据质控结果
[0123][0124]
注:读段数量:左右两端均比对上的且在合理距离范围内的读段数目。umi重复率:按照比对至同一位点,片段长度一致且umi一致的片段仅保留一条的方式去除重复读段的比例。坐标重复率:按照比对至同一位点且片段长度一致的片段仅保留一条的方式去除重复读段的比例。umi拯救率:考虑umi后“拯救”的读段占去除的所有重复(不考虑umi)读段的比例。frip:计算落在读段富集区域中的读段比例。tss score:计算tss周围(上下游3000bp)读段最高富集倍数。
[0125]
通过实施例可以获得7个水稻样品atac-seq信息,在混库测序后,成功拆分得到7个品种的测序读段(如表3)。将umi标签进行分类,发现通过重复插入事件造成的重复序列占比情况如表4。
[0126]
结果表明,umi-atac-seq可以挽救标准atac-seq中约20%被误认为pcr重复的读段(或约占总比对到基因组的读段的6%)。进一步将基因组分成250个bp的bins,并计算每个bin中的被挽救的读段数。发现染色质上tn5越容易接近的区域,通过umi方法可以挽救的读段数就越多。在样本c019中,81.8%被挽救的读段数来自水稻基因组中最易接近的前2.0%的bins。并且,被挽救的读段数在易受拷贝数变异或基因组组装错误影响的区域中富集,例如rdna区域、转座子、逆转录转座子和端粒区(图5)。
[0127]
为了评估加入umi对定量的影响,分别通过基于相同基因组坐标进行的pcr重复数据消除(cd)、基于umi的重复数据消除(ud)和不进行重复数据消除(nd)来量化每个250bp bin的tn5插入,并相互比较。结果表明,ud和nd的相关性略高于ud和cd(r
2
分别为0.995和0.961);并得到了与其他六个样品相似的结果(图6a、b)。随着染色质区域可及性的增加,cd的量化更偏离ud(图6c)。这些结果表明,在atac-seq的定量分析中,仅基于比对坐标的pcr重复的去除可能比不去除重复结果更糟糕。
[0128]
使用pydnase4对通过cd和ud鉴定到的转录因子足迹进行进一步的验证。大多数足迹都是在高度易接近的区域被识别出来的,因此在去除pcr重复的方法中受到的影响更大。ud识别的足迹数量明显大于cd(图7a)。虽然只有约6%的读段被挽救,但它们却促成了50%或更多的足迹被发现。比较重叠足迹的得分,发现ud计算的足迹得分几乎总是高于cd(图7b)。
[0129]
对于仅由cd或ud识别的足迹,使用相同的足迹区间计算ud和cd的足迹得分。结果
表明,在大多数情况下,ud足迹得分也高于cd。使用igv5(整合基因组学观察者)检查已识别足迹周围tn5插入的频率,发现足迹样分布在ud中更为明显(图7c)。对于仅限于ud的足迹,许多独立的tn5插入作为pcr重复被移除,导致cd无法识别。此外,足迹深度(fpd)值越大,可以解释为足迹区的tn5插入量较其侧翼区减少(图7d)。通过计算fpd值,结果显示ud的fpd值普遍高于cd,说明ud模式下足迹模式更为明显(图7e)。因此,umi-atac-seq可以在染色质高度可及的区域挽救了大量独立的tn5插入事件,从而提高转录因子足迹绘制的灵敏度和准确性。
[0130]
其它未详细说明的部分均为现有技术。尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施例,而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他实施例,这些实施例都属于本发明保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips