
 商标分类
商标分类  商标转让
商标转让 
一种环状硫酸酯的制备方法与流程
 2021-02-02 12:02:56|
2021-02-02 12:02:56| 531|
531| 起点商标网
起点商标网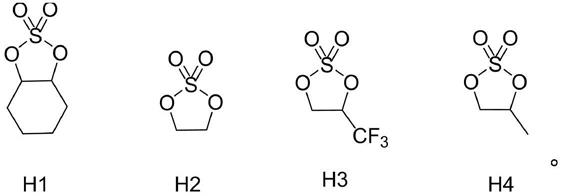
[0001]
本发明涉及有机合成领域,尤其涉及一种环状硫酸酯的制备方法。
背景技术:
[0002]
环状硫酸酯类材料早就为人所知,且它在有机合成中一直受到极大的关注。近年来,有大量文献介绍具有这种类似结构的物质作为医药和表面活性剂的中间体,具有广阔的应用前景。近年来,环状硫酸酯类材料逐渐作为锂离子电池电解液添加剂使用,能够有效抑制电极表面的副反应。
[0003]
目前(如中国专利cn109485633、cn109369609、cn108707095、cn102241662)该类化合物的主要合成路线如下:
[0004][0005]
该路线需两步反应,首先使用二醇化合物和氯化亚砜反应得到亚硫酸酯,进一步在贵金属三氯化钌催化下,采用次氯酸钠氧化得到目标物。
[0006]
另外,中国专利申请号cn201910393379.x公开了以乙二醇和氯化亚砜为原料,合成亚硫酸酯,然后将亚硫酸乙烯酯在催化剂(氯化钯配合物催化剂和氯化铜催化剂)的作用下与空气或氧气发生氧化反应生成硫酸乙烯酯,经过滤、水洗浓缩结晶等步骤,得到硫酸乙烯酯成品。
[0007]
该路线的主要问题在于:(1)使用氯化亚砜,产生大量的腐蚀性气体氯化氢;(2)第二步氧化反应需使用三氯化钌作为催化剂,该催化剂价格昂贵,且不易回收套用,同时使用次氯酸钠作为氧化剂,反应放热剧烈,不易控制,能耗较高;(3)使用次氯酸钠作为氧化剂产生较大量的含盐废水,增大了废水处理成本。即使专利cn201910393379.x改用空气或氧气作为氧化剂,但该反应必须在氯化钯和氯化铜类催化剂存在下才能发生,该类催化剂价格昂贵,特别是最近几年含钯催化剂价格增加4倍,且该工艺催化剂回收困难,不符合绿色化学的发展要求。
技术实现要素:
[0008]
本发明针对现有合成环状硫酸酯工艺中酸性气体量大,设备腐蚀严重、废水量大,且盐含量大、反应放热不易控制等问题,提供一种环状硫酸酯的制备方法,其反应式为:
[0009][0010]
原料1中,r
1
和r
2
分别独立地选自自氢、甲基、三氟甲基或r
1
与r
2
链接成环;原料2
中,x选自氢或nh
2
;酸酐选自醋酸酐或丙酸酐。
[0011]
基于上述机理,本发明的操作可为:将原料1、原料2和酸酐加入到反应容器中进行反应,反应温度0℃-150℃,反应时间10min-24h,反应完毕,得到环状硫酸酯反应液;向其加入冰水和卤代烷烃,搅拌打浆,分层、水洗,将所得有机相减压脱溶剂,采用卤代烷烃和低碳烷烃混合溶剂重结晶,即得。
[0012]
在上述操作中,原料2用量摩尔数为原料1的0.9-5倍,优选1-3倍;酸酐用量摩尔数为原料1的1-5倍,优选2.5-4倍;卤代烷烃选自二氯乙烷、二氯甲烷或氯仿中的一种;低碳烷烃选自正己烷、环己烷、石油醚、正庚烷或正辛烷中的一种。
[0013]
本发明环状硫酸酯具有如式h1-h4所示的结构:
[0014][0015]
本发明的有益效果是:本发明提供了一种采用环氧化合物作为原材料,在酸酐参与下,与氨基磺酸或硫酸反应一步制得环状硫酸酯,制备过程无需贵金属催化,无腐蚀性气体的产出,所得产物纯度高度,色度低(<20hazen),水分含量≤20ppm,酸值≤10ppm,有效改变了电解液中水分和酸值对电池循环性能和储存稳定性的影响。另外,本发明所提供的反应路线中所涉及原料均为大宗工业品,廉价易得,可大幅降低产物原材料成本。
具体实施方式
[0016]
以下结合实例对本发明进行描述,所举实例只用于解释本发明,并非用于限定本发明的范围。
[0017]
实施例1
[0018]
化合物h1的制备:称取98.1g(1.0mol)环氧环己烷,106.8g(1.1mol)氨基磺酸和224.6g(2.2mol)醋酸酐加入到1000ml三口烧瓶中,磁力搅拌,n
2
(10ml/min)保护,控内温120℃,与此温下保温反应4h,降温至-5-0℃,将反应物缓慢倾倒入冰水中,同时加入300g二氯乙烷搅拌1.0h,分层、有机相水洗,干燥,减压脱溶剂至无馏分,进一步通过减压蒸馏(128-135℃,50-100pa)纯化得精品150.1g,收率84.23%,gc纯度99.92%,色度8hazen。gc-ms:178,
1
h nmr(400mhz):溶剂氘代氯仿,δ(ppm):2.602-0.919ppm(m,8h),5.792-5.493(m,2h)。
[0019]
实施例2
[0020]
化合物h2的制备:将1l高压釜预冷至-10℃,向釜中加入44.0g(1.0mol)环氧乙烷,339.8g(3.5mol)氨基磺酸和357.3g(3.5mol)醋酸酐,机械搅拌,控内温10-20℃,保温反应24.0h,停止反应,降温至0-5℃,缓慢转移至冰水中,加入400g二氯甲烷,搅拌1.0h,分层,有机相水洗,氧化铝干燥,减压脱溶剂至体系稍有固体析出;控内温30-35℃,向体系中加入缓慢230g正庚烷,体系成白色浑浊状,于此温下打浆搅拌30min,降温至0-5℃,抽滤得白色固体,进一步通过减压干燥得精品90.8g,收率73.22%,gc纯度99.86%,色度11hazen,熔点
(dsc):98.0-99.1-99.8℃。gc-ms:124,
1
hnmr(400mhz):溶剂氘代氯仿,δ(ppm):4.731ppm(s,4h)。
[0021]
实施例3
[0022]
化合物h3的制备:将1l高压釜预冷至-10℃,向釜中加入112.0g(1.0mol)三氟甲基环氧丙烷,缓慢加入150g(1.5mol)浓硫酸和306.3g(3.0mol)醋酸酐,机械搅拌,控内温30-40℃,保温反应8.0h,停止反应,降温至0-5℃,缓慢转移至冰水中,加入400g二氯甲烷,搅拌1.0h,分层,有机相水洗,氧化铝干燥,减压脱溶剂至无馏分,进一步通过减压蒸馏(主馏分52-56℃,250-300pa)得精品147.8g,收率76.93%,gc纯度99.90%,色度9hazen。gc-ms:192,
1
h nmr(400mhz):溶剂氘代氯仿,δ(ppm):5.149-5.131(l h,m),4.912-4.888(1h.dd),4.802-4.778(lh,dd)。
[0023]
实施例4
[0024]
化合物h4的制备:将1l高压釜预冷至-10℃,向釜中加入58.0g(1.0mol)甲基环氧丙烷,缓慢加入150g(2.0mol)浓硫酸和255.3g(2.5mol)醋酸酐,机械搅拌,控内温30-40℃,保温反应9.0h,停止反应,降温至0-5℃,缓慢转移至冰水中,加入600g二氯甲烷,搅拌1.0h,分层,有机相水洗,氧化铝干燥,减压脱溶剂至体系稍有固体析出;控内温30-35℃,向体系中加入缓慢220g正庚烷,体系成白色浑浊状,于此温下打浆搅拌30min,降温至0-5℃,抽滤得白色固体,进一步通过减压干燥得精品104.2g,收率75.43%,gc纯度99.91%,色度13hazen,熔点(dsc):57.4-58.2-59.7℃。gc-ms:138,
1
h nmr(400mhz):溶剂氘代氯仿,δ(ppm):5.181-5.141ppm(m,1h),4.732-4.711ppm(dd,1h),4.310-4.289ppm(dd,1h),1.596-1.580(d,3h)。
[0025]
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签: 硫酸

 热门咨询
热门咨询




tips