
 商标分类
商标分类  商标转让
商标转让 
靶向蛋白降解c-Met降解剂及其制备方法与应用与流程
 2021-02-02 11:02:23|
2021-02-02 11:02:23| 445|
445| 起点商标网
起点商标网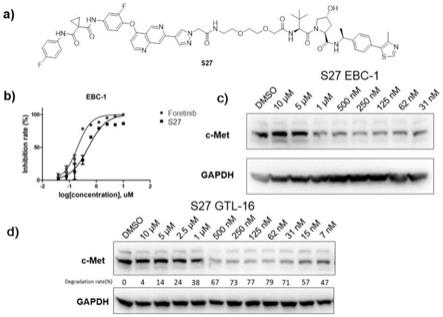
靶向蛋白降解c-met降解剂及其制备方法与应用
技术领域
[0001]
本发明涉及一种靶向蛋白降解c-met降解剂及其制备方法与应用,属于医药技术领域。
背景技术:
[0002]
c-met又称肝细胞生长因子(hgf)受体,属于受体酪氨酸激酶家族(rtks)。hgf与c-met结合可以调控下游的多种信号通路参与多种生理过程,包括细胞的增殖和存活、细胞凋亡和血管生成等;当hgf/c-met信号通路发生异常时就会导致肿瘤的发生。此外,c-met信号通路的过表达与临床上较差的愈后和egfr抑制剂的耐药有关。克唑替尼(crizotinib)和卡博替尼(cabozantinib)是目前已经上市的两个c-met小分子抑制剂,临床肿瘤治疗效果很好,但是都面临用药后获得性耐药降低疗效的难题。目前临床上报道的c-met的耐药机制主要有基因的扩增和过表达、c-met突变以及旁通路激活如c-myc的上调等。因此,针对耐药机制引入新的手段和新的技术逆转c-met获得性耐药,是临床分子靶向肿瘤治疗的迫切需求。
[0003]
靶向蛋白降解嵌合体protac(proteolysis targeting chimeras)是一种基于细胞自身的泛素蛋白酶体系统发展而来的新型靶向降解目标蛋白的技术。protac分子是一种双功能的分子,既能与目标蛋白结合,又能招募e3泛素连接酶,从而将目标蛋白泛素化,进而通过蛋白酶体将目标蛋白降解。protac分子相比于传统的小分子抑制剂有几个明显的优势:首先protac分子可以降解整个目标蛋白,以激酶为例,小分子抑制剂只能够抑制激酶的酶活活性,而protac分子可以降解整个激酶,包括激酶的非酶活功能。因此protac分子更接近于基因表型上的敲降;其次protac分子可以作为一种催化剂,protac分子降解目标蛋白后可以重新释放进而结合新的目标蛋白,因此protac可能仅需要非常低的剂量就能发挥作用;最后,蛋白的重新合成需要一定的时间,因此protac分子的作用时间可能相对更持久。
技术实现要素:
[0004]
本发明的主要内容是针对于c-met小分子抑制剂获得性耐药的问题,提供一种基于靶向蛋白降解protac策略的c-met降解剂及其制备方法和应用,以及该类c-met降解剂在治疗非小细胞肺癌和胃癌等癌症中的应用。
[0005]
本发明的第一个目的是提供一种结构式如式i所示的化合物,其光学异构体,及药学上可接受的盐或溶剂合物,
[0006]
m-l-e
[0007]
式i
[0008]
其中:
[0009]
m表示c-met激酶的配体,l表示连接链,e表示e3泛素连接酶的配体;
[0010]
m为式ii-1或式ii-2所示化合物,
[0011][0012]
其中,cy
1
为iii所示化合物或不存在,cy
1
结构位ar环与吡啶的2,3位的碳原子发生并环,并且ar环选自苯环,c
5-6
芳基,5-6个环原子组成的杂芳基;
[0013][0014]
l为式-所示化合物以下任意结构之一或者不存在:
[0015][0016]
其中,各个n表示独立的1-10之间的任意一个整数;
[0017]
e为式v所示化合物的任意结构之一:
[0018][0019]
其中,cy
2
为-ch
2-、-nh-、-o-、不存在或式vi所示化合物的任意结构之一;r
1
表示-ch
3
、-ch
2
ch
3
、-och
3
、或者不存在;
[0020][0021]
vi所示化合物中,各个x表示-ch
2-、-nh-或-o-,y表示羰基或-ch
2-,z表示n或c原子,r
1
表示-ch
3
、-ch
2
ch
3
、-och
3
、或者不存在。
[0022]
进一步地,式i所示化合物的结构式为s1-s36任意结构之一,
[0023]
[0024]
[0025][0026]
本发明的第二个目的是提供一种靶向蛋白降解c-met降解剂,包括所述结构式如式i所示的化合物,其光学异构体,及药学上可接受的盐或溶剂合物。
[0027]
本发明的第三个目的是提供所述靶向蛋白降解c-met降解剂,以及与其他药物联合用药用于制备治疗或预防肿瘤疾病的药物中的应用。
[0028]
进一步地,所述肿瘤疾病为肺癌、乳腺癌、结肠癌、前列腺癌、胰腺癌、肝癌、卵巢癌、急性骨髓性白血病、多发性骨髓瘤、肾癌、胃癌中的一种或几种。
[0029]
进一步地,所述其他药物包括依鲁替尼、环磷酰胺、多柔吡星、阿糖胞苷、azacitidine、decitabine、卡非佐米、沙利度胺、来那度胺、泊马渡胺、吉非替尼、厄洛替尼、奥司他丁、阿法替尼、氟他胺、尼鲁米特中的至少一种。
[0030]
本发明的有益效果:
[0031]
本发明的化合物具有显著的c-met降解作用与细胞增殖抑制作用,具有作为抗肿瘤药物治疗肿瘤的潜力,并且本发明化合物在慢病毒转染构建的ebc-1耐药细胞株中表现出显著的增殖抑制活性,且明显优于小分子抑制剂lxm-262,并且显著提高了细胞选择性,说明本发明实例化合物在克服肿瘤c-met获得性耐药性上具有显著的优势,尤其是化合物s27只对c-met依赖的ebc-1肺癌细胞表现出较好的活性,说明化合物s27具有较好的细胞选择性,而原本的小分子抑制剂是多靶点的,对c-met非依赖的细胞系也有抑制效果。
附图说明
[0032]
图1是化合物s9、s15和s16在ebc-1细胞中化合物不同浓度下的c-met降解作用;
[0033]
图2是化合物s19的结构、化合物对ebc-1肿瘤细胞的增殖抑制活性以及ebc-1细胞中化合物不同浓度下的c-met降解作用;
[0034]
图3是化合物s27的结构、化合物s27对ebc-1肿瘤细胞的增殖抑制活性以及化合物s27分别在ebc-1和gtl-16肿瘤细胞中不同浓度下的c-met降解作用;
[0035]
图4是阴性对照化合物s28的结构、化合物s28对ebc-1肿瘤细胞的增殖抑制活性以
及化合物s28在ebc-1肿瘤细胞中不同浓度下的c-met降解作用;
[0036]
图5是验证化合物s27降解c-met通过泛素-蛋白酶体途径;
[0037]
图6是测试化合物s27在浓度250nm条件下对gtl-16肿瘤细胞中c-met降解的时间依赖性;
[0038]
图7是化合物s36的结构以及化合物s36分别在ebc-1和gtl-16肿瘤细胞中不同浓度下的c-met降解作用;
[0039]
图8是化合物s27、s28以及lxm-262在不同ebc-1耐药细胞株中的增殖抑制作用;
[0040]
图9是化合物s36以及lxm-262在不同ebc-1耐药细胞株中的增殖抑制作用;
[0041]
图10是化合物s27以及lxm-262在不同肺癌细胞株中的抗增殖抑制作用。
具体实施方式
[0042]
不需进一步详细说明,认为本领域熟练技术人员借助前面的描述,可以最大程度地利用本发明。因此,下面提供的实施例仅仅是进一步阐明本发明而己,并不意味着以任何方式限制本发明范围。
[0043]
原料可以从商业途径获得,或者通过本领域已知的方法制备,或根据本文所述方法制备。化合物的结构通过核磁共振(
1
h-nmr)和/或质谱(ms)来确定。nmr测定是用varian inova(300mhz)或者bruker advance(400mhz)核磁共振仪,测定溶剂为氘代氯仿(cdcl
3
)、氘代二甲亚砜(dmso-d
6
),tms为内标。ms的测定用waters uplc-mass spectrometer或者agilent 1100lc/msd trap sl version mass spectrometer(esi)液相色谱-质谱联用仪。柱层析采用青岛海洋化工厂的200-300目硅胶。
[0044]
实施例1:制备2-(4-(4-(2-氟-4-(1-(((4-氟苯基)氨基甲酰基)环丙烷-1-甲酰胺基)苯氧基)-1,6-萘啶-7-基)-1h-吡唑-1-基)乙酸
[0045][0046]
以上反应路线中,所使用的试剂与条件如下:a)2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯,n,n-二异丙基乙胺,n,n-二甲基甲酰胺;b)氢气,10%湿钯碳,乙醇;c)碳酸铯,n,n-二甲基甲酰胺,110℃;d)四(三苯基膦)钯,碳酸铯,1,4-二氧六环:水(5:
1),120℃;e)溴乙酸叔丁酯,碳酸钾,n,n-二甲基甲酰胺;f)三氟乙酸,二氯甲烷。反应步骤具体如下:
[0047]
步骤1:n-(4-(苄氧基)-3-氟苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺
[0048][0049]
将1-((4-氟苯基)氨基甲酰基)环丙烷-1-羧酸(2.9g,13mmol),4-(苄氧基)-3-氟苯胺(2.82g,13mmol)溶于10ml n,n-二甲基甲酰胺,加入n,n-二异丙基乙胺(5g,39mmol)和2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(5.4g,14.3mmol),常温搅拌反应过夜。反应完成后加入乙酸乙酯(200ml)稀释,加入饱和碳酸氢钠水溶液(50ml*3)洗涤,饱和食盐水(50ml*3)洗涤。乙酸乙酯相无水硫酸钠干燥,过滤,旋干,经柱层析纯化(石油醚:乙酸乙酯=4:1)得白色固体(4.7g,产率85%)。
1
h nmr(400mhz,cdcl
3
)δ8.94(s,1h),8.90(s,1h),7.50
–
7.29(m,8h),7.05
–
7.01(m,3h),6.96
–
6.91(m,1h),5.12(s,2h),1.63(s,4h).ms(esi,[m+h]
+
)m/z 423.18.
[0050]
步骤2:n-(3-氟-4-羟基苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺
[0051][0052]
将n-(4-(苄氧基)-3-氟苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(3.65g,8.65mmol)溶于100ml甲醇中,加入10%湿钯碳(365mg,10%wt),置换氢气后,常温搅拌3小时。反应完成后,过滤,滤液旋干,经柱层析纯化(石油醚:乙酸乙酯=5:1至3:1)得白色固体(2.08g,产率72%)。
1
h nmr(400mhz,dmso-d
6
)δ10.06(s,1h),9.91(s,1h),9.61(s,1h),7.62(dd,j=8.8,5.2hz,2h),7.52(d,j=13.2hz,1h),7.16
–
7.12(m,3h),6.86(t,j=9.4hz,1h),1.42(s,4h).ms(esi,[m+h]
+
)m/z 333.36.
[0053]
步骤3:n-(4-((7-氯-1,6-萘啶-4-基)氧基)-3-氟苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺
[0054][0055]
将n-(3-氟-4-羟基苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(606mg,1.82mmol),4,7-二氯-1,6-萘啶(330mg,1.66mmol)溶于5ml n,n-二甲基甲酰胺,加入碳酸铯(1.08g,3.32mmol)。置换氩气后,110℃搅拌反应2小时。反应完成后加入乙酸乙酯(200ml)稀释,加入饱和碳酸氢钠水溶液(50ml*3)洗涤,饱和食盐水(50ml*3)洗涤。乙酸乙酯相无水硫酸钠干燥,过滤,旋干,经柱层析纯化(石油醚:乙酸乙酯=4:1至1:1)得白色固体(718mg,产率88%)。
1
h nmr(400mhz,cdcl
3
)δ10.32(s,1h),9.60(s,1h),8.82(d,j=
5.6hz,1h),8.02(s,1h),7.97(s,1h),7.84
–
7.80(m,1h),7.46
–
7.42(m,2h),7.33
–
7.23(m,2h),7.08(t,j=8.4hz,1h),6.56(d,j=5.2hz,1h),1.65
–
1.61(m,4h).ms(esi,[m+h]
+
)m/z 495.21.
[0056]
步骤4:n-(4-((7-(1h-吡唑-4-基)-1,6-萘啶-4-基)氧基)-3-氟苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺
[0057][0058]
将n-(4-((7-氯-1,6-萘啶-4-基)氧基)-3-氟苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(555mg,1.12mmol),4-吡唑硼酸频哪醇酯(654mg,3.37mmol)溶于1,4-二氧六环:水(5:1)的混合溶剂,加入碳酸铯(733mg,2.25mml),四(三苯基膦)钯(116mg,0.11mmol)。置换氩气后,120℃搅拌反应3小时。反应完成后,过滤,滤液加入乙酸乙酯(200ml)萃取,加入饱和碳酸氢钠水溶液(50ml*3)洗涤,饱和食盐水(50ml*3)洗涤。乙酸乙酯相无水硫酸钠干燥,过滤,旋干,经柱层析纯化(石油醚:乙酸乙酯=4:1至1:1)得淡黄色固体(511mg,产率87%)。
1
h nmr(400mhz,dmso-d
6
)δ13.20(s,1h),10.46(s,1h),10.03(s,1h),9.69(s,1h),8.85(d,j=5.2hz,1h),8.55(s,1h),8.28(s,1h),8.22(s,1h),7.96(dd,j=13.2,2.4hz,1h),7.66(dd,j=9.2,5.2hz,2h),7.60
–
7.49(m,2h),7.17(t,j=8.8hz,2h),6.62(d,j=5.2hz,1h),1.56
–
1.46(m,4h).ms(esi,[m+h]
+
)m/z 527.41.
[0059]
步骤5:叔丁基2-(4-(4-(2-氟-4-(1-(((4-氟苯基)氨基甲酰基)环丙烷-1-羧酰胺基)苯氧基)-1,6-萘啶-7-基)-1h-吡唑-1-基)乙酸酯
[0060][0061]
将n-(4-((7-(1h-吡唑-4-基)-1,6-萘啶-4-基)氧基)-3-氟苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(526mg,1mmol)溶于2ml n,n-二甲基甲酰胺,加入碳酸钾(207mg,1.5mmol),溴乙酸叔丁酯(195mg,1mmol),常温搅拌反应3小时。反应完成后,加入100ml乙酸乙酯稀释。乙酸乙酯相加入饱和碳酸氢钠水溶液(50ml*3)洗涤,饱和食盐水(50ml*3)洗涤。乙酸乙酯相无水硫酸钠干燥,过滤,旋干,经柱层析纯化(二氯甲烷:甲醇=100:1至100:4)得淡黄色固体(500mg,产率78%)。.
1
h nmr(400mhz,cdcl
3
)δ10.19(s,1h),9.73(s,1h),8.77(d,j=5.2hz,1h),8.19
–
8.15(m,3h),8.02(s,1h),7.80(d,j=11.6hz,1h),7.47
–
7.43(m,2h),7.32
–
7.25(m,2h),7.07(t,j=8.4hz,2h),6.49(d,j=5.2hz,1h),4.90(s,2h),1.85
–
1.81(m,2h),1.64
–
1.60(m,2h),1.50(s,9h).ms(esi,[m+h]
+
)m/z 640.7.
[0062]
步骤6:2-(4-(4-(2-氟-4-(1-(((4-氟苯基)氨基甲酰基)环丙烷-1-甲酰胺基)苯氧基)-1,6-萘啶-7-基)-1h-吡唑-1-基)乙酸
[0063][0064]
将叔丁基2-(4-(4-(2-氟-4-(1-(((4-氟苯基)氨基甲酰基)环丙烷-1-羧酰胺基)苯氧基)-1,6-萘啶-7-基)-1h-吡唑-1-基)乙酸盐(500mg,0.78mmol)溶于4ml二氯甲烷,缓慢加入三氟乙酸(2ml),常温搅拌过夜。反应完成后,旋干溶剂得黄色油状物(460mg,粗品)。直接进行下一步反应,不需要任何纯化。
1
h nmr(400mhz,dmso-d
6
)δ10.45(s,1h),10.01(s,1h),9.71(s,1h),8.88(d,j=5.2hz,1h),8.52(s,1h),8.26(s,1h),8.18(s,1h),7.95(d,j=13.2hz,1h),7.67
–
7.60(m,2h),7.57
–
7.50(m,2h),7.16(t,j=8.8hz,2h),6.67(d,j=5.6hz,1h),5.06(s,2h),1.51
–
1.44(m,4h).ms(esi,[m+h]
+
)m/z 585.49.
[0065]
实施例2:制备2-((2-(2,6-二氧杂哌啶-3-基)-1,3-二氧异吲哚啉-4-基)氧基)乙酸
[0066][0067]
试剂与条件:a)吡啶,3-氨基-2,6-哌啶二酮盐酸盐,110℃;b)溴乙酸叔丁酯,碳酸钾,n,n-二甲基甲酰胺;c)三氟乙酸,二氯甲烷。
[0068]
步骤1:2-(2,6-二氧代哌啶-3-基)-4-羟基异吲哚啉-1,3-二酮
[0069][0070]
将4-羟基异苯并呋喃-1,3-二酮(952mg,5.84mmol)溶于10ml吡啶,加入3-氨基-2,6-哌啶二酮盐酸盐(960mg,5.84mmol),反应液加热到110℃搅拌反应过夜。反应完成后冷却至室温,减压蒸馏除去吡啶,剩余物经柱层析纯化(二氯甲烷:甲醇=100:1至100:3)得白色固体(1.5g,产率93%)。
1
h nmr(400mhz,dmso-d
6
)δ11.20(s,1h),11.11(s,1h),7.66(t,j=7.8hz,1h),7.32(d,j=7.2hz,1h),7.25(d,j=8.4hz,1h),5.08(dd,j=12.8,5.2hz,1h),2.95-2.83(m,1h),2.64
–
2.45(m,2h),2.06
–
1.98(m,1h).
[0071]
步骤2:叔丁基2-((2-(2,6-二氧代哌啶-3-基)-1,3-二氧代异吲哚-4-基)氧基)乙酸酯
[0072][0073]
将2-(2,6-二氧代哌啶-3-基)-4-羟基异吲哚啉-1,3-二酮(500mg,1.8mmol)溶于4ml n,n-二甲基甲酰胺,加入碳酸钾(378mg,2.7mmol),溴乙酸叔丁酯(356mg,1.8mmol)。常温搅拌反应2小时,反应完成后加入100ml乙酸乙酯稀释。乙酸乙酯相加入饱和碳酸氢钠水溶液(50ml*3)洗涤,饱和食盐水(50ml*3)洗涤。乙酸乙酯相无水硫酸钠干燥,过滤,旋干,经柱层析纯化(石油醚:乙酸乙酯=1:1)得白色固体(640mg,产率92%)。
1
h nmr(400mhz,cdcl
3
)δ8.02(s,1h),7.68(t,j=7.8hz,1h),7.52(d,j=7.2hz,1h),7.11(d,j=8.4hz,1h),4.97(dd,j=12.0,5.2hz,1h),4.79(s,2h),2.95
–
2.70(m,3h),2.18
–
2.10(m,1h),1.48(s,9h).ms(esi,[m+h]
+
)m/z 411.36.
[0074]
步骤3:2-((2-(2,6-二氧杂哌啶-3-基)-1,3-二氧异吲哚啉-4-基)氧基)乙酸
[0075][0076]
将叔丁基2-((2-(2,6-二氧代哌啶-3-基)-1,3-二氧代异吲哚-4-基)氧基)乙酸酯(300mg,0.77mmol)溶于4ml二氯甲烷,缓慢加入三氟乙酸(2ml),常温搅拌反应4小时。反应完成后,旋干溶剂得白色固体(260mg,粗品)。直接进行下一步反应,不需要任何纯化。
1
h nmr(400mhz,dmso-d
6
)δ13.28(br,1h),11.13(s,1h),7.80(t,j=8.0hz,1h),7.48(d,j=7.2hz,1h),7.40(d,j=8.4hz,1h),5.76(s,1h),5.11(dd,j=12.8,5.6hz,1h),5.00(s,2h),2.95
–
2.84(m,1h),2.63
–
2.51(m,2h),2.08
–
2.00(m,1h).ms(esi,[m+h]
+
)m/z 333.28.
[0077]
实施例3:
[0078][0079]
试剂与条件:a)2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯,n,n-二异丙基乙胺,n,n-二甲基甲酰胺;b)三氟乙酸,二氯甲烷;c)2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯,n,n-二异丙基乙胺,n,n-二甲基甲酰胺。
[0080]
步骤1:叔丁基(2-(2-((2-(2,6-二氧代哌啶-3-基)-1,3-二氧异吲哚基-4-基)氧基)乙酰氨基)乙基)氨基甲酸酯
[0081][0082]
将2-((2-(2,6-二氧杂哌啶-3-基)-1,3-二氧异吲哚啉-4-基)氧基)乙酸(1eq),(2-氨基乙基)氨基甲酸叔丁酯(1.2eq)溶于2ml n,n-二甲基甲酰胺,常温搅拌下加入n,n-二异丙基乙胺(3eq)和2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(1.1eq),继续搅拌反应1小时,反应完成后,加入50ml乙酸乙酯稀释,乙酸乙酯相加入饱和碳酸氢钠水溶液(50ml*3)洗涤,饱和食盐水(50ml*3)洗涤。乙酸乙酯相无水硫酸钠干燥,过滤,旋干,经柱层析纯化(二氯甲烷:甲醇=100:1至100:5)得白色固体(产率95%)。
1
h nmr(400mhz,cdcl
3
)δ8.46(s,1h),7.78-7.69(m,2h),7.57(d,j=7.2hz,1h),7.22(d,j=8.4hz,1h),5.09
–
5.02(m,1h),5.01
–
4.95(m,1h),4.72
–
4.60(m,2h),3.53
–
3.45(m,2h),3.36
–
3.29(m,2h),2.94
–
2.89(m,1h),2.81
–
2.71(m,2h),2.22
–
2.13(m,1h),1.41(s,9h).ms(esi,[m+h]
+
)m/z 496.6.
[0083]
步骤2:n-(2-氨基乙基)-2-((2-(2,6-二氧代哌啶-3-基)-1,3-二氧异吲哚基-4-基)氧基)乙酰胺三氟乙酸盐
[0084][0085]
将叔丁基(2-(2-((2-(2,6-二氧代哌啶-3-基)-1,3-二氧异吲哚基-4-基)氧基)乙酰氨基)乙基)氨基甲酸酯(1eq)溶于4ml二氯甲烷,缓慢加入三氟乙酸(2ml),常温搅拌反应4小时。反应完成后,旋干溶剂得白色固体。直接进行下一步反应,不需要任何纯化。
[0086]
步骤3:n-(4-((7-(1-(2-((2-(2-((2-(2,6-二氧代哌啶-3-基)-1,3-二氧异吲哚-4-基)氧基)乙酰胺基)乙基)氨基)-2-氧乙基)-1h-吡唑-4-基)-1,6-萘啶-4-基)氧基)-3-氟苯基)-n-(4-氟苯基)环丙烷-1,1-二甲酰胺(s1)
[0087][0088]
将2-(4-(4-(2-氟-4-(1-(((4-氟苯基)氨基甲酰基)环丙烷-1-甲酰胺基)苯氧基)-1,6-萘啶-7-基)-1h-吡唑-1-基)乙酸(1eq),n-(2-氨基乙基)-2-((2-(2,6-二氧代哌啶-3-基)-1,3-二氧异吲哚基-4-基)氧基)乙酰胺三氟乙酸盐(1.1eq)和n,n-二异丙基乙胺
(3eq)溶于2ml n,n-二甲基甲酰胺中,常温搅拌5分钟后加入2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(1.1eq),继续搅拌反应4小时。反应完成后,加入50ml乙酸乙酯稀释,乙酸乙酯相加入饱和碳酸氢钠水溶液(50ml*3)洗涤,饱和食盐水(50ml*3)洗涤。乙酸乙酯相无水硫酸钠干燥,过滤,旋干,经柱层析纯化(二氯甲烷:甲醇=100:3至100:8)得白色固体(产率24%)。
1
h nmr(400mhz,dmso-d
6
)δ11.12(s,1h),10.44(s,1h),10.02(s,1h),9.66(s,1h),8.83(d,j=5.2hz,1h),8.46(s,1h),8.32
–
8.25(m,1h),8.23(s,1h),8.16(s,1h),8.14
–
8.09(m,1h),7.94(d,j=13.2hz,1h),7.80(t,j=7.8hz,1h),7.69
–
7.60(m,2h),7.58
–
7.45(m,3h),7.40(d,j=8.8hz,1h),7.16(t,j=8.8hz,2h),6.60(d,j=5.2hz,1h),5.13(dd,j=12.8,5.2hz,1h),4.86(s,2h),4.79(s,2h),3.27
–
3.18(m,4h),2.95
–
2.82(m,1h),2.64
–
2.53(m,2h),2.09
–
1.96(m,1h),1.55
–
1.41(m,4h).ms(esi,[m+h]
+
)m/z 941.2.
[0089]
按照实施例1-3的步骤,分别制备下列化合物s2-s36:
[0090]
[0091]
[0092][0093]
实施例4:
[0094]
htrf kinease c-met激酶活性抑制实验
[0095]
表1化合物对c-met的酶活抑制活性结果
[0096]
[0097][0098]
nt表示not tested,没有测试
[0099]
本发明实施例化合物对c-met激酶酶活抑制作用如下:
[0100]
如表1所示,实施例化合物的c-met酶活抑制活性均在纳摩尔级别,其中部分化合物的ic
50
基本与小分子c-met抑制剂母体化合物lxm-262和阳性对照foretinib相当,甚至部分化合物优于阳性对照。表明本专利实施例化合物保留了较强的c-met酶活抑制活性。
[0101]
实施例5:
[0102]
cck-8法测定化合物对非小细胞肺癌细胞系ebc-1的细胞增殖抑制活性
[0103]
表2化合物对c-met依赖的非小细胞肺癌细胞系ebc-1的抗增殖活性
[0104]
[0105][0106]
本发明实施例化合物对c-met依赖的非小细胞肺癌细胞系ebc-1的抗增殖活性:
[0107]
如表2所示,实施例化合物对肿瘤细胞ebc-1均表现出明显的抗增殖活性,其中部分化合物的抗增殖活性显著优于lxm-262和foretinib。其中化合物s36的活性相比于母体化合物lxm-262提升了30倍。综上所述,本发明的实施例化合物具有显著的抗肿瘤细胞增殖作用,并且优于母体小分子和阳性对照。
[0108]
实施例6:
[0109]
western-blot实验测定化合物对c-met激酶的降解作用
[0110]
本发明实施例化合物对c-met激酶的降解作用如下:
[0111]
如图1所示,从western-blot实验中可以观察到,在ebc-1细胞系中实例化合物s9和s16具有显著的c-met降解作用,分别在7nm和30nm时表现出最优的降解活性相比于dmso空白对照组。化合物s15没有降解活性。
[0112]
如图2所示,在western-blot实验结果中可以观察到,实例化合物s19在浓度为7nm时相比于dmso空白对照组表现出了微弱的降解作用。
[0113]
如图3所示,从western-blot实验结果中可以观察到,实例化合物s27在ebc-1细胞系中表现出了显著的降解活性。在浓度低至31nm时相比于dmso空白对照组表现出明显的c-met降解作用,随着给药浓度增加,降解作用逐渐增强,并在250nm时达到最大的c-met蛋白
降解作用。随着浓度的进一步增大,降解作用会逐步降低。在胃癌细胞系gtl-16中,化合物s27同样表现出显著的c-met降解作用。具体表现为当给药浓度为7nm时,化合物s27相比于dmso空白对照组表现出显著的降解作用,随着浓度的逐渐增加,降解作用逐渐增强,在浓度为62nm时达到最大降解作用。随着给药浓度的进一步增加,化合物的降解作用逐渐下降。
[0114]
如图4所示,实例化合物s28是化合物s27的阴性对照化合物。具体表现为化合物s28对ebc-1肿瘤细胞的增殖抑制作用相较于s27减弱了10倍。在western-blot实验中可以观察到,化合物s27不具有c-met降解作用,说明实例化合物s27的细胞活性来源于化合物的蛋白降解作用。
[0115]
如图5所示,在western-blot实验结果中可以观察到,实例化合物s27在浓度为250nm时表现出显著的c-met降解作用。c-met小分子抑制剂lxm-262在浓度为250nm和1μm时均没有c-met降解作用,并且e3泛素连接酶vhl的小分子配体在浓度为1μm时同样没有c-met降解作用。使用nedd8抑制剂mln4924和蛋白酶体抑制剂carfilzomib均能抑制实例化合物s27的蛋白降解作用,说明化合物s27降解c-met通过泛素-蛋白酶体途径。
[0116]
如图6所示,从western-blot结果中可以观察到,实例化合物s27在gtl-16细胞系中,当浓度为250nm时,在给药后的2小时相比于dmso空白对照组表现出显著的c-met降解作用,随着时间的进一步延长,在12小时的时候仍然表现出显著的降解作用。
[0117]
如图7所示,从western-blot结果中可以观察到,实例化合物s36在ebc-1和gtl-16细胞系中均没有表现出c-met降解作用。
[0118]
综上所述,本发明实例化合物具有显著的c-met降解作用与细胞增殖抑制作用,表明本发明的实例化合物具有作为抗肿瘤药物治疗肿瘤的潜力。
[0119]
实施例7:
[0120]
慢病毒转染ebc-1耐药细胞株的构建与cck-8法测定化合物对ebc-1耐药细胞株的增殖抑制活性
[0121]
本发明实例化合物对ebc-1耐药细胞株的增殖抑制活性如下:
[0122]
如图8所示,实例化合物s27对ebc-1
tpr-met
、ebc-1
l1157t
和ebc-1
d1228y
三株耐药细胞株均表现出显著的抗增殖活性,且均优于小分子抑制剂lxm-262。阴性对照化合物s28相比于化合物s27和小分子抑制剂lxm-262表现出较差的细胞活性。
[0123]
如图9所示,实例化合物s36对ebc-1
tpr-met
、ebc-1
l1157t
和ebc-1
d1228y
三株耐药细胞株均表现出显著的抗增殖活性,且均优于小分子抑制剂lxm-262。
[0124]
如图10所示,实例化合物s27对多种非小细胞肺癌肺癌的抗增殖活性,实验表明化合物s27只对c-met依赖的ebc-1肺癌细胞表现出较好的活性,说明化合物s27具有较好的细胞选择性。
[0125]
综上所述,本发明实例化合物在慢病毒转染构建的ebc-1耐药细胞株中表现出显著的增殖抑制活性,且明显优于小分子抑制剂lxm-262,并且显著提高了细胞选择性,说明本发明实例化合物在克服肿瘤c-met获得性耐药性上具有显著的优势。
[0126]
以上所述仅是本发明的优选实施方式,并不用于限制本发明,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明技术原理的前提下,还可以做出若干改进和变型,这些改进和变型也应视为本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips