
 商标分类
商标分类  商标转让
商标转让 
用于敲低PD1基因表达的microRNA及其嵌合抗原受体-T/NK细胞的构建的制作方法
 2021-02-02 11:02:52|
2021-02-02 11:02:52| 416|
416| 起点商标网
起点商标网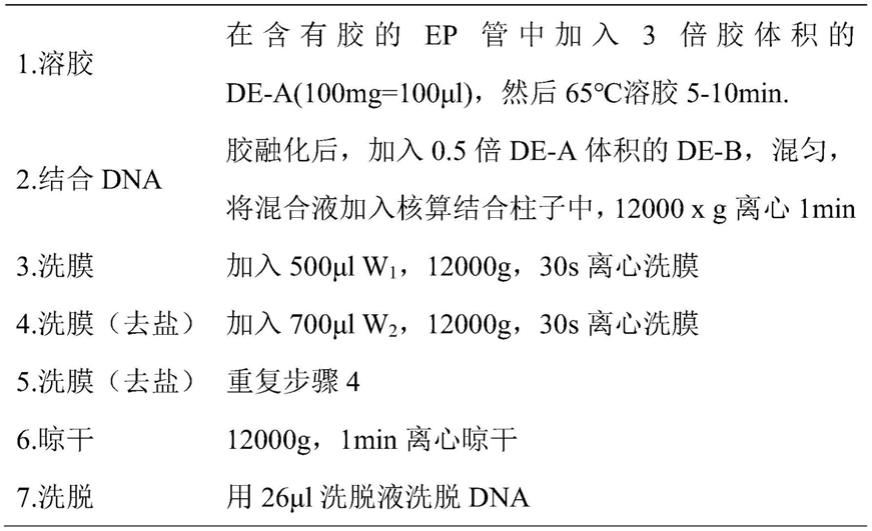
用于敲低pd1基因表达的microrna及其嵌合抗原受体-t/nk细胞的构建
技术领域
[0001]
本发明属于生物工程技术领域,具体涉及一种用于高效敲低pd1基因表达的microrna及其嵌合抗原受体-t/nk细胞的构建。
背景技术:
[0002]
car-t/nk疗法是一种以嵌合型抗原受体为基础的细胞免疫治疗方案。通过体外基因转移技术,将编码嵌合抗原受体(car)的基因序列转导入t/nk细胞中,生成可以结合靶抗原的肿瘤特异性t/nk细胞。近几年,cart细胞免疫治疗在血液肿瘤的临床前和临床试验中表现出巨大的潜力,但由于肿瘤微环境中免疫检查点的抑制作用,cart细胞在实体瘤治疗中的效果一直不尽如人意。
[0003]
pd-1(程序性死亡受体)是一种表达于t细胞表面的蛋白质分子,当pd-1与其配体pd-l1或pd-l2结合时可抑制下游nf-κb基因的转录,抑制干扰素-γ的分泌,从而抑制t细胞的杀伤活性。其中pd-1/pd-l1通路已经被证实与t细胞耗竭有关,会导致cart细胞功能减退,肿瘤清除能力受损等问题。采用pd-1抗体或pd-l1抗体阻断pd-1与pd-l1、pd-l2的结合,可使t细胞的肿瘤杀伤活性不受抑制,从而提高t细胞杀肿瘤的效果。
[0004]
为了解决上述问题,发明专利201711196439.6公开了一种敲除pd1的靶向cd20的嵌合抗原受体t细胞及其制备方法和应用,采用crispr/cas9敲除cart细胞pd-1,虽然该方法表现出了高效抗肿瘤效果,然而也有研究发现在初始cd8+t细胞中程序性细胞死亡因子1基因(pdcd1)的意外缺失会增加其耗竭并损伤细胞的存活性和功能,并且基因敲除会对其它基因产生不可预期的破坏和影响。
[0005]
rna干扰(rna interference,rnai)是指在进化过程中高度保守的、由双链rna(double-stranded rna,dsrna)诱发的、通过抑制mrna翻译或使mrna高效特异性降解从而介导转录后的基因沉默现象。由dsrna介导的基因沉默过程通常需要经过三个步骤来实现。首先是dsrna进入细胞后与酶蛋白dicer结合并被dicer切割成21-23nt长的小片段,这些小片段被称为sirna;紧接着sirna再与体内一些酶(包括内切酶、外切酶、解旋酶等)结合形成rna诱导的沉默复合物(rna-induced silencing complex,risc);最后被激活的risc在反义链的引导下与目标mrna结合,通过抑制mrna的翻译或使mrna降解介导转录后的基因沉默。所以基因沉默是一种能降低目的基因表达的分子机制,它不像基因敲除会消除整个目的基因的表达,只会使靶基因的表达降低至少70%。与传统的基因敲除手段相比,rnai技术具有快速、高效、特异性强、操作简易等优点,近年来已经成为揭示不同物种中基因功能的工具,被广泛应用于植物、线虫、果蝇及哺乳动物。所以我们假设,如果使用sirna的方式使cart细胞中的pd-1表达降低,这样t/nk细胞不仅能躲避肿瘤微环境中的抑制信号,并且还避免完全敲除pd1基因带来的损害和且存在脱靶风险。
技术实现要素:
[0006]
针对现有技术的上述不足,本发明所要解决的技术问题是:如何提供一种用于敲低pd1基因表达的microrna及其嵌合抗原受体-t/nk细胞的构建,解决肿瘤细胞存在逃逸问题及基因敲除存在脱靶风险的问题。
[0007]
为了解决上述技术问题,本发明采用了如下的技术方案:一种用于高效敲低pd1基因表达的microrna,所述microrna由seq id no.1或seq id no.2所示的核苷酸序列编码而成。
[0008]
本发明的另一个目的在于,提供一种重组载体,含有编码所述microrna的核苷酸序列。
[0009]
本发明的另一个目的在于,提供一种重组慢病毒,将含有编码所述microrna的核苷酸序列的重组质粒及辅助质粒共同转染宿主细胞,得到所述重组慢病毒。
[0010]
作为优选的,所述慢病毒包装所用宿主细胞为293t细胞。
[0011]
本发明的另一个目的在于,提供一种嵌合抗原受体-t/nk细胞,所述编码嵌合抗原受体(car)结构包括第一信号、第二信号和第三信号;所述第一信号为抗原结合结构域,所述第二信号为跨膜结构域和细胞内传导结构域,所述第三信号为权利要求1所述的microrna,且所述microrna插入在ef1α启动子内。这样,编码microrna的核苷酸序列位于慢病毒载体ef1α启动子的下游,则因干扰了宿主细胞内源性microrna的转运和加工、会导致慢病毒载体包装滴度显著下降。
[0012]
本发明的另一个目的在于,提供所述嵌合抗原受体-t/nk细胞的构建方法,将所述重组慢病毒转染阳性t/nk淋巴细胞中,培养后收获细胞,从而制得所述能敲低内源性pd-1表达的嵌合抗原受体-t/nk细胞。
[0013]
进一步,所述阳性t淋巴细胞是从人源外周血单个核细胞中分离获得。
[0014]
本发明的另一个目的在于,提供一种细胞抑制剂(t/nk杀伤效应细胞),其有效成分为所述嵌合抗原受体-t/nk细胞。
[0015]
本发明的另一个目的在于,提供所述细胞抑制剂在制备抗肿瘤药物中的应用。
附图说明
[0016]
图1为plenti-anti-cd19(pd1 knockdown)质粒示意图。
[0017]
图2为microrna插入ef1a启动子的位置对病毒包装滴度的影响。
[0018]
图3为靶向pd1的microrna对car-t细胞中pd1表达量的影响。
[0019]
图4为敲低pd1的car-t细胞对表达gfp的靶细胞raji的杀伤活性;a为流式细胞术分析图,b为柱状分析图。
具体实施方式
[0020]
下面结合具体实施例和附图对本发明作进一步详细说明。以下实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。
[0021]
主要实验材料:ecor v-hf、mlui-hf、ndei限制性内切酶(neb公司),无缝克隆酶(和元生物),高保真prime gxl star酶(takara公司),transstbl3感受态细胞(全式金生物科技有限公司),plasmid mini kit i(omega),plasmid maxi kit(qiagen),
dmem、rpmi-1640、opti-mem培养基、gibco fbs(thermo fisher scientific),sanger测序(上海桑尼生物有限公司),nacl、酵母粉、蛋白胨、edta、naoh(上海生工生物工程股份有限公司),引物(江苏金唯智生物科技有限公司)。
[0022]
一、敲低内源pd1基因表达的嵌合抗原受体-t/nk细胞的构建(1)构建plenti-cd19-microrna#pd1重组质粒:
[0023]
靶向pd1的microrna(seq id no.1、seq id no.2)由苏州金唯智生物科技有限公司合成,使用ndel和mlui-hf双酶切plenti-ef1a-anti-cd19car载体,使其双方均带有ndel和mlui粘性末端,反应条件为37℃3h,65℃20min,酶切体系如表3。酶切产物经过1%琼脂糖凝胶电泳获得载体片段,然后用xygene胶回收试剂盒回收plenti载体片段以及靶向pd1的microrna(操作步骤见下表1),检测浓度和纯度。载体片段和目的片段通过t4克隆(体系如表2),16℃16-24h后,65℃10min,然后进行质粒转化(t4克隆产物在冰上放置5min后,将其转入50μl transstbl3感受态中,冰上放置30min,42℃45s,再冰上5min,加入500μl lb,在37℃,225rpm/min摇床中活化1h,然后5000rpm/min 20℃离心5min,弃去上清,将剩余菌液混匀后涂板,37℃培养12-14小时)。挑单克隆菌落进行菌液扩增37℃,250rpm/min,12h-14h,质粒提取,最终用aflii-hf限制性内切酶酶切鉴定,最后进行sanger测序,筛选阳性克隆。成功构建如图1所示的plenti-cd19-microrna#pd1重组质粒,其中,将seq id no.1插入ef1α启动子内,记为plenti-cd19-mirna-#pd1#1;seq id no.2插入ef1α启动子内,记为plenti-cd19-mirna-#pd1#2。
[0024]
表1.胶回收
[0025][0026]
表2.t4克隆体系
[0027][0028]
表3.限制性酶切体系
[0029][0030][0031]
(2)重组慢病毒构建
[0032]
s1:在15cm细胞皿中培养293t细胞,等待293t细胞长满至全视野70%时,用1.5ml pbs重悬60μgpei(pbs-pei),1.5ml pbs重悬总质量为20μg的plenti-cd19-microrna#pd1及辅助质粒(pbs-dna);然后将pbs-pei混合液加至pbs-dna得到pei-dna-pbs混合液中,室温静置20min。
[0033]
s2:准备opti-dmem全培于37℃培养箱中复温,吸弃293t细胞中的dmem原培养基,将opti-dmem沿皿壁加入到293t细胞;然后将pei-dna-pbs混合液加至培养皿中,37℃培养48h;
[0034]
s3:收集上清液中的慢病毒于50ml离心管,再加20ml培养基孵育24h,以收集72h内的病毒,1500rpm离心5min去除细胞碎片,或用针筒通过0.45μm过滤器过滤后,3000
×
g离心12-14h 4℃,以浓缩病毒。
[0035]
s4:离心结束后,弃去上清,尽量去除残留在管壁上的液体,以1:200-1:400加入vivo全培或aim-v全培(最好加1%hepes),轻轻反复吹打重悬病毒,将病毒分装于1.5ml ep管中,保存于-80℃,避免反复冻融(冻融使滴度降低一个数量级),得到重组慢病毒。
[0036]
(3)敲低pd1表达量的嵌合抗原受体t细胞的构建
[0037]
s1:pbmc(外周血单个核细胞)的分离
[0038]
取健康人外周血10ml至edta-na2抗凝管,与dpbs按照1:1的比例混匀;取四只15ml
无菌离心管,分别加入5ml ficoll分离液,将外周血与dpbs的混合液缓慢加至ficoll分离液面上,注意不要破坏液面,水平离心800g,20min,25℃,加减速度均调到“0”,离心后用巴氏吸管将离心管中的白色絮状层即pbmc层吸出,置于新的无菌离心管中,加入pbs,将pbmc离心洗两遍,1500rpm/min,5min水平离心,弃掉上清,加入1ml buffer1(dpbs含5%fbs)重悬计数pbmc,得到pbmc。
[0039]
s2:免疫磁珠法分离抗原特异性t淋巴细胞
[0040]
用流式细胞术确定pbmc中cd3阳性细胞的比例。在细胞悬液中以cd3/cd28 dynabeads:cd3阳性细胞=3:1的比例加入cd3/cd28 beads(106个cd3阳性细胞加30μl beads),4℃以1rpm速度旋转摇晃30min使磁株与细胞充分接触结合;30分钟以后,在试管中加足够(大于1ml)buffer1,然后把试管放于磁力架上左右旋转1~2分钟,吸弃上清;配制vivo完全培养基:vivo空培+5%fbs+1%hepes+1%丙酮酸钠+1%非必需氨基酸+1:30谷氨酰胺+1:10000il-2+1:2000il-7+1:2000il-15,并用vivo全培重悬细胞与磁珠,计数;加培养基使cd3阳性细胞浓度在0.5-1x106/ml之间。铺板细胞浓度为0.5~1.0
×
106/ml,置于5%co
2 37℃培养箱培养24-36h,采用磁铁对孵育好磁珠的细胞进行筛选;pbs洗涤,去除免疫磁珠后,得到cd3阳性t淋巴细胞。
[0041]
s3:病毒转染法制备抗原特异性t淋巴细胞
[0042]
以moi值=5、20、40、80将重组慢病毒转导经过免疫磁珠分离法得到的cd3阳性t淋巴细胞,moi(病毒感染复数)=[病毒滴度x病毒体积(ml)]/细胞数;1200xg,90min,4℃离心后,在37℃培养箱孵育至96孔板长满细胞后转至24孔板中,5-7天时测car转导率,收集细胞,得到敲低内源pd1的嵌合抗原受体t细胞。
[0043]
二、性能验证
[0044]
1、将包装出的慢病毒转染jurkat cell以计算病毒滴度。
[0045]
将jurkat细胞1500rpm离心5min后,弃上清,1ml 1640培养基重悬,计数;然后于96孔板中加入0.5x10
6 jurkat细胞,以1:50、1:500、1:1000、1:2000等梯度比例加入病毒,再补充培养基至每孔总体积为200μl;每孔加入0.1μl polybreneb蛋白促转导(0.1μl/200μl体系);将96孔板经1200g,90min,32℃离心,离心后,于37℃培养箱孵育4h;再将96孔板各孔的jurkat细胞悬液吹打混匀后转至1.5mlep管中,1500rpm离心5min后,弃上清,用1ml 1640全培重悬后转至24孔板中扩大培养48h,37℃。结果如图2所示。
[0046]
从图中可以看出,分别靶向pd1的如seq id no.1与seq id no.2所示序列插入ef1a启动子内,得到anti-cd19(mirna-#pd1#1)car-t与anti-cd19(mirna-#pd1#2)car-t病毒感染jurkat细胞,表达均在80%左右,与不带有mirna的病毒感染jurkat细胞(anti-cd19car-t)能力相当,说明包装滴度没有明显的变化。但如seq id no.2所述序列插入ef1a启动子后方得到的病毒(anti-cd19car-t-mirna#pd1#2),感染jurkat细胞仅有60%的表达,与不带mirna的病毒有明显的差异,说明microrna插入ef1a启动子内,病毒包装滴度并不受影响。
[0047]
2、利用流式细胞仪测定上述敲除pd1的靶向cd19的嵌合抗原受体t细胞的pd1表达量。
[0048]
取转导慢病毒后的t细胞,脱磁处理,2500rpm离心3min,弃去上清;然后用facs重悬细胞,2500rpm离心3min,弃去上清,再加入流式抗体,避光,孵育20min后,用facs洗两次,
流式上机分析细胞表面pd1表达情况,结果如图3所示。
[0049]
从图中可以看出,与不表达靶向pd1的mirna的car-t细胞相比,表达mirna#pd1#1与mirna#pd1#2的病毒,car-t细胞表达pd1的量明显降低,且具有高效的敲低率,可以达到85%以上,所以证明我们筛选的两条针对pd1高效敲除的mirna序列。
[0050]
3、通过流式细胞术检测敲低pd1的嵌合抗原受体t细胞的体外肿瘤细胞杀伤情况。
[0051]
培养raji-luc-gfp细胞至对数生长状态,取一定细胞数离心沉淀后计数;在96孔平底不透明白板中加入10
4
个raji-luc-gfp细胞,培养基补充至100μl;分别将anti-cd19car-t、anti-cd19(microrna#pd1#1)car-t、anti-cd19(microrna#pd1#2)car-t细胞与raji-luc-gfp细胞数比例设定为5:1、10:1、20:1,将相应的car-t细胞加入每孔混合培养;设置mock细胞组,t细胞的数目与上述car-t细胞数相同;同时设置两个对照,阴性对照为raji-luc-gfp细胞在培养基中培养;阳性对照为在培养基加入2.5%的triton-x 100,两者均不加mock细胞或car-t细胞,作为细胞杀伤的最小和最大化背景值,即kmin和kmax。培养4小时后,96孔板以1500rpm速度离心5min,弃掉上清,用培养基洗一次后重悬细胞,流式细胞表面染色后,检测杀伤结果。结果如图4所示。
[0052]
从图中可以看出,分别将anti-cd19 car-t与靶细胞共孵育后,与对照组相比,anti-cd19(microrna#pd1#1)car-t和anti-cd19(microrna#pd1#2)car-t孵育液中靶细胞仅剩余1.22%和3.07%,比原来增强了约4倍。说明在ef1α启动子内插入seq id no.1或seq id no.2能显著增强anti-cd19 car-t对肿瘤细胞的杀伤力。并且由于mirna设计的靶向位置不同,可能对pd1的敲低效率不同,导致杀伤情况不同。由以上结果表明经本发明方法制备的敲低pd1基因表达后的的靶向cd19的嵌合抗原受体t细胞具有高效且特异性的肿瘤杀伤能力,可避免肿瘤细胞逃脱免疫监视。
[0053]
以上仅是本发明优选的实施方式,需指出的是,对于本领域技术人员在不脱离本技术方案的前提下,作出的若干变形和改进的技术方案应同样视为落入本权利要求书要求保护的范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips