
 商标分类
商标分类  商标转让
商标转让 
一种4-氟吡啶-2-胺的制备方法与流程
 2021-02-02 11:02:54|
2021-02-02 11:02:54| 338|
338| 起点商标网
起点商标网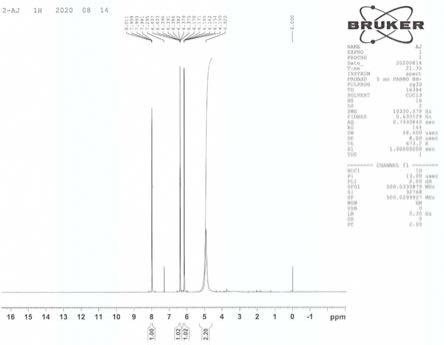
[0001]
本发明涉及一种4-氟吡啶-2-胺的制备方法。
背景技术:
[0002]
4-氟吡啶-2-胺是一种新型的含氟医药中间体,是制备pi3k抑制剂、ruat-1抑制剂、酪氨酸激酶抑制剂、醛固酮合酶抑制剂等抑制剂的重要中间体,在医药领域有广泛的应用。目前4-氟吡啶-2-胺的合成方法主要有以下3种:方法一(wo2011079076;wo2012082689;wo2014009295),以2-氯-4-氟吡啶、氨基甲酸叔丁酯为原料,经钯催化偶联得到4-氟吡啶-2-氨基甲酸叔丁酯,然后使用三氟乙酸脱boc保护基得到4-氟吡啶-2-胺。该方法使用的原料2-氯-4-氟吡啶和催化剂价格昂贵,成本高,不适合大规模工业化生产。合成路线如下式所示:。
[0003]
方法二(cn107759515a),以2-氨基吡啶为原料,经n-氧化反应,硝化反应、氨基的酰化保护反应、还原反应、重氮化反应、席曼反应、氨基脱保护反应得到4-氟吡啶-2-胺。该方法合成路线长、操作复杂、三废量大、污染严重、收率低、成本高,不适合工业化生产。合成路线如下式所示:。
[0004]
方法三(cn108440402a;广东化工,2013,17(40):229),以2-吡啶甲酸为原料经二氯亚砜氯代和酰氯的氨化得到中间体4-氯吡啶—2甲酰胺,或以4-氯吡啶盐酸盐和甲酰胺为原料经minisci反应制备得到中间体4-氯吡啶-2-甲酰胺。然后4-氯吡啶-2-甲酰胺经霍夫曼降解、卤素交换反应得到4-氟吡啶-2-胺。该方法最后一步使用4-氯吡啶-2-胺为原料进行卤素交换反应制备4-氟吡啶-2-胺。按照文献和专利的实验方法和条件,使用4-氯吡啶-2-胺和氟化钾为原料进行多次验证实验,均未得到产物4-氟吡啶-2-胺。反应机理分析如下:芳香化合物卤素交换反应的机理是亲核取代反应机理,只有与缺电子芳环相连的卤原子才能发生卤素交换反应,且必须在非质子的极性溶剂中进行反应。4-氯吡啶-2-胺的吡啶环的2-位为富电子的氨基基团,氨基氮原子上的孤对电子与吡啶环形成p-π共轭,增加了吡啶环的电子云密度,导致吡啶环的缺电子程度极度弱化,严重钝化4-氯吡啶-2-胺底物上
氯原子的活性。从反应原理上分析,4-氯吡啶-2-胺很难发生卤素交换反应,该路线的卤素交换反应存在严重的可行性问题。合成路线如下式所示:。
技术实现要素:
[0005]
本发明的目的是提供一种成本低、原料易得,反应条件温和,操作简便、产品质量好,适合工业化生产的4-氟吡啶-2-胺制备方法。为达到上述发明目的,本发明所提供的4-氟吡啶-2-胺的制备方法,包括以下步骤:(1)以4-氯吡啶-2-甲腈为原料,在催化剂催化下与氟盐进行卤素交换反应制备得到4-氟吡啶-2-甲腈;(2)将4-氟吡啶-2-甲腈在过氧化氢促进下水解得到4-氟吡啶-2-甲酰胺;(3)4-氟吡啶-2-甲酰胺进行霍夫曼降解反应得到4-氟吡啶-2-胺。
[0006]
所述步骤(1)中卤素交换反应的具体方式为:将4-氯吡啶-2-甲腈、氟盐和催化剂加入到交换反应溶剂中,在温度为120~160℃下进行反应2~5小时,反应完毕后将反应液倒入水中,再加入乙酸乙酯进行萃取,萃取完毕收集有机层,脱除有机层中的乙酸乙酯,得到4-氟吡啶-2-甲腈。
[0007]
所述氟盐为氟化钾、氟化钠、氟化钙和氟化铯中的任意一种或二种以上的混合物;所述反应溶剂为二甲基亚砜、二甲基砜、n,n'-二甲基甲酰胺、n-甲基吡咯烷酮和n,n'-二甲基乙酰胺中的任意一种或二种以上的混合物;催化剂为四丁基溴化铵、四丁基氯化铵、四苯基溴化鏻和四乙基溴化铵中的任意一种或二种以上的混合物;所述4-氯吡啶-2-甲腈与氟盐的摩尔比为1:1.1~3.0;4-氯吡啶-2-甲腈与催化剂的摩尔比为1:0.05~0.2。反应溶剂的用量以将4-氯吡啶-2-甲腈完全溶解为底限。
[0008]
所述步骤(2)中的水解具体方式为:将4-氟吡啶-2-甲腈溶入有机溶剂中,加入氢氧化钠水溶液,然后滴加过氧化氢水溶液,滴加完毕后在温度30~60℃下反应3~5小时,反应完毕后固液分离得到4-氟吡啶-2-甲酰胺。
[0009]
所述有机溶剂为乙醇、丙酮和二甲基亚砜中的任意一种,有机溶剂的用量以将4-氟吡啶-2-甲腈完全溶解为底限;所述氢氧化钠水溶液的浓度为1~10%;4-氟吡啶-2-甲腈与过氧化氢的摩尔比为1:1.0~2.0;4-氟吡啶-2-甲腈与氢氧化钠的摩尔比为1:0.1~0.5。
[0010]
所述步骤(3)中霍夫曼降解反应的具体方式为:将氢氧化钠溶入水,在温度0~10℃下滴加溴素,滴加完毕,在温度0~10℃下继续保温反应1~1.5小时,然后加入4-氟吡啶-2-甲酰胺,在温度10~80℃下反应3~10小时,反应完毕后加入亚硫酸钠水溶液除去未反应
140.0[m
+
]。
[0018]
(3)在1l三口瓶安装温度计及套管、球形冷凝管、机械搅拌,向三口瓶中加入159.2g水,68.0g(1.7mol)氢氧化钠,待氢氧化钠溶解后,将反应瓶转移至低温浴槽中,降温至0~10℃,滴加67.1g(0.42mol)溴素,滴加时间为15-30分钟,滴毕,在0~10℃条件下保温反应1~1.5小时;然后加入56.0g(0.4mol)上述步骤(2)制备的4-氟吡啶-2-甲酰胺,并缓慢升温至60~80℃反应6~8小时,反应完毕,加入亚硫酸钠水溶液除去未反应的溴,搅拌30min后抽滤,收集滤饼,使用甲苯对滤饼重结晶,重结晶完毕进行固液分离得到4-氟吡啶-2-胺,收率82.6%,gc纯度99.3%。质谱分析:ms(ei) m/z: 112.0[m
+
]。
[0019]
实施例24-氟吡啶-2-胺的制备方法包括以下步骤:(1)在1l三口瓶安装n2侧管、温度计及套管、球形冷凝管、液封、机械搅拌、干燥管。向三口瓶中依次加入69.3g(0.50mol)4-氯吡啶-2-甲腈,10.5g(0.025mol)四苯基溴化鏻,63.0g(1.50mol)干燥氟化钠,346.5g二甲基亚砜,氮气保护,机械搅拌,升温至140~160℃保温反应3~5小时,反应完毕,将反应液降至室温后倒入692g水中,然后加入400g乙酸乙酯进行萃取,萃取完毕收集有机层,减压蒸馏除去有机层中的乙酸乙酯,得到4-氟吡啶-2-甲腈,收率92.8%,gc纯度95.6%;(2)在1l三口瓶安装温度计及套管、球形冷凝管、机械搅拌,向三口瓶中依次加入56.2g(0.46mol)上述步骤(1)制备的4-氟吡啶-2-甲腈、80g丙酮、92g氢氧化钠水溶液(氢氧化钠质量浓度10.0%),称取102.0g(0.9mol)质量浓度30%的双氧水,并转移至恒压滴液漏斗中,室温搅拌条件下将双氧水缓慢滴入上述1l三口反应瓶中,滴加时间为15-30分钟,滴加完毕升温至30~40℃,并保温反应3~4小时,反应结束后将反应液降至室温后抽滤得到4-氟吡啶-2-甲酰胺,收率86.2%,gc纯度99.3%。
[0020]
(3)在1l三口瓶安装温度计及套管、球形冷凝管、机械搅拌,向1l三口瓶中加入360g水、80.0g(2.0mol)氢氧化钠、待氢氧化钠溶解后,将反应瓶转移至低温浴槽中,降温至0~10℃,滴加83.1g(0.52mol)溴素,滴加时间为15-30分钟;滴加完毕,在0~10℃条件下保温反应1~1.5小时;将56.0g(0.4mol)上述步骤(2)制备的4-氟吡啶-2-甲酰胺加入上述反应瓶中,并缓慢升温至40~60℃反应8~10小时;反应完毕,加入亚硫酸钠水溶液除去未反应的溴,搅拌30min后抽滤,收集滤饼,使用甲苯对滤饼进行重结晶,重结晶完毕进行固液分离得到4-氟吡啶-2-胺,收率80.3%,gc纯度99.6%。
[0021]
实施例34-氟吡啶-2-胺的制备方法包括以下步骤:(1)在1l三口瓶安装n2侧管、温度计及套管、球形冷凝管、液封、机械搅拌、干燥管。向1l三口瓶中依次加入69.3g(0.50mol)2-氰基-4-氯吡啶、8.3g(0.03mol)四丁基氯化铵、12.6g(0.06mol)四乙基溴化铵、91.1g(0.60mol)干燥氟化铯、150g二甲基砜、196g n-甲基吡咯烷酮,氮气保护,机械搅拌,升温至130~150℃保温反应2~3小时,反应完毕,将反应液降至室温后倒入692g水中,然后加入400g乙酸乙酯进行萃取,萃取完毕收集有机层,减压蒸馏除去有机层中的乙酸乙酯,得到4-氟吡啶-2-甲腈,收率93.1%,gc纯度95.8%;(2)在1l三口瓶安装温度计及套管、球形冷凝管、机械搅拌,向1l三口瓶中依次加入56.2g(0.46mol)上述步骤(1)制备的4-氟吡啶-2-甲腈、80g二甲基亚砜、122g氢氧化钠水溶
液(氢氧化钠质量浓度5.0%),称取79.4g(0.7mol)质量浓度30%的双氧水,并转移至恒压滴液漏斗中,室温和搅拌条件下将双氧水缓慢滴入上述1l三口反应瓶中,滴加时间为15-30分钟;滴加完毕升温至30~50℃,并保温反应3~5小时,反应结束,将反应液降至室温后抽滤得到4-氟吡啶-2-甲酰胺,收率86.7%,gc纯度98.9%。
[0022]
(3)在1l三口瓶安装温度计及套管,球形冷凝管、机械搅拌,向1l三口瓶中加入864g水,96.0g(2.4mol)氢氧化钠,待氢氧化钠溶解后,将反应瓶转移至低温浴槽中,降温至0~10℃,滴加121.4g(0.76mol)溴素,滴加时间为15-30分钟;滴加完毕,在0~10℃条件下保温反应1~1.5小时;将56.0g(0.4mol)上述步骤(2)制备的4-氟吡啶-2-甲酰胺加入上述反应瓶中,并缓慢升温至30~50℃反应3~6小时;反应完毕,加入亚硫酸钠水溶液除去未反应的溴,搅拌30min后抽滤,收集滤饼,使用甲苯对滤饼进行重结晶,重结晶结束后固液分离得到4-氟吡啶-2-胺,收率81.8%,gc纯度99.5%。
[0023]
图1是实施例1制备的产品的
1
h nmr图,其表征的内容如下:
1
h nmr (cdcl
3
, 500 mhz),δ: 7.98~8.01 (dd, 1h),6.38~6.41 (m, 1h),6.15~6.18 (dd, 1h),4.92 (s, 2h)。
[0024]
根据质谱和核磁氢谱分析,实施例1的产品为4-氟吡啶-2-胺,其结构式如下:。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips