
 商标分类
商标分类  商标转让
商标转让 
一种AMG837及手性γ-甲基苯戊醇的合成方法与流程
 2021-02-02 10:02:30|
2021-02-02 10:02:30| 304|
304| 起点商标网
起点商标网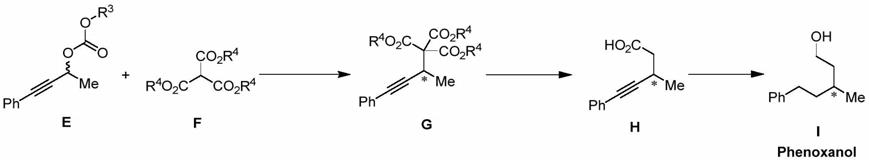
一种amg837及手性
γ-甲基苯戊醇的合成方法
技术领域
[0001]
本发明属于不对称有机合成技术领域,具体涉及一种amg837及手性γ-甲基苯戊醇的合成方法。
背景技术:
[0002]
当今社会,随着经济的快速发展以及人们生活水平的提高,人类健康正面临着各种疾病的威胁。其中,全球目前有接近3亿的糖尿病患者,并且还在以较快的速度不断增加,2型糖尿病(t2dm)的流行给全世界的医疗保健系统带来了巨大压力。2型糖尿病表现为多种代谢缺陷,其中最突出的是身体组织对胰岛素作用的抵抗力增强,以及胰腺对葡萄糖负荷的胰岛素分泌反应显着降低。针对这两个缺陷中的后者,磺酰脲类胰岛素促分泌剂抑制了胰岛β细胞中存在的atp敏感性钾通道,并引起胰岛素释放的持续升高。但这种增加胰岛素分泌的机制对血浆葡萄糖水平不敏感,并可能导致过度释放,导致低血糖症,并伴有出汗,神经质,头晕和神志不清的症状。而游离脂肪酸增加胰岛素分泌的能力已经在体外和体内进行了广泛的研究。通过鉴定,gpr40作为胰岛表达的游离脂肪酸受体,可以促进胰岛β细胞钙离子内流,分泌胰岛素,同时起到抑制血糖升高的作用。值得注意的是,gpr40引起的胰岛素分泌增加,仅在葡萄糖水平升高的时候存在。而amg837作为gpr40激动剂,可以通过激活gpr40 受体实现控制血糖的目的,因此开发amg837的合成方法具有非常广泛的药用价值。
[0003]
也正是因为它令人感兴趣的生物学特性,因此amg837的不对称合成研究在商业和科学上都引起了广泛的关注。目前为止,关于amg837的合成方法已经有若干条路线的报道 (cn110590767、org.lett.2011,13,952.、angew.chem.int.ed.2013,52,7532.、bioorg.med. chem.lett.2012,22,1267.),但是这些方法存在合成路线较长、产率较低、对映选择性较差、需要进行手性拆分或较难实现放大量制备等问题,因此,探索从简单易得的消旋分子骨架出发,以立体选择性方式针对性的快速制备amg837的新型合成路线是非常有必要的。
[0004]
γ-甲基苯戊醇又名玫瑰花醇,是常用的玫瑰香系列香精香料。也正是由于其非同寻常的香气以及持久性,因此在高档的日用品及护肤品中得以广泛的应用。而众所周知,在构型相反的化合物之间,气味阈值会存在显着的差异,因此手性γ-甲基苯戊醇的合成在香精香料的应用上具有广泛的商业前景。但是,由于缺乏有效的手性γ-甲基苯戊醇对映选择性合成方法,因此目前市场主要使用消旋的γ-甲基苯戊醇作为香料添加剂。尽管目前关于手性γ-甲基苯戊醇的合成已有相关报道(chirality2011,23,761.、acs catal.2016,6,8342.、org.lett.2019,21, 3247.),但是由于存在合成路线较长、产率较低或较难实现工业化制备等缺点,开发以立体选择性方式快速制备手性γ-甲基苯戊醇的新型合成路线是非常有必要的。
技术实现要素:
[0005]
本发明的目的是提供一种amg837及手性γ-甲基苯戊醇的合成方法,从而解决现
有技术中存在的上述问题。
[0006]
本发明amg837的合成方法,包括如下步骤:
[0007]
步骤1:氩气氛围下,将双-(1,5-环辛二烯)镍金属催化剂与手性膦配体在有机溶剂中混合预搅15分钟,随后在氩气保护下,将消旋的炔丙基碳酸酯化合物a、甲烷三甲酸酯化合物b、碱加入到混合体系中,在室温至50℃条件下反应12~96小时,薄层色谱点板确定反应终点;随后将该反应体系用乙酸乙酯稀释,并加水萃取,然后将水相用乙酸乙酯萃取3次,合并有机相后用无水硫酸钠干燥并减压浓缩,然后以石油醚/乙酸乙酯(4:1,v/v)作为洗脱剂,通过柱层析分离得到不对称炔丙基化产物c;
[0008]
步骤2:空气环境下,将不对称炔丙基化产物c置于有机溶剂中搅拌,并向反应体系中加入氢氧化钠水溶液(3n),随后将混合物在室温至80℃下搅拌,并通过tlc监测;当反应体系中没有起始原料时,真空除去挥发物,向反应体系中加入乙酸并加热至回流条件下反应 6~12小时;待反应结束后,将乙酸真空浓缩,将该反应体系用乙酸乙酯稀释并加水萃取,然后将水相用乙酸乙酯萃取3次,合并有机相后用无水硫酸钠干燥并减压浓缩,然后以石油醚/乙酸乙酯(1:1,v/v)作为洗脱剂,通过柱层析分离得到amg837或其对映异构体。
[0009]
步骤1中,双-(1,5-环辛二烯)镍金属催化剂、手性膦配体与甲烷三甲酸酯化合物b的摩尔比为0.1:0.12:1;甲烷三甲酸酯化合物b与消旋的炔丙基碳酸酯化合物a的摩尔当量比为1:1~3;甲烷三甲酸酯化合物b与碱的摩尔比为1:1~3。
[0010]
上述amg837制备过程的反应路线如下:
[0011][0012]
式中:*代表手性碳原子;取代基r
1
选自甲基、乙基、叔丁基;取代基r
2
选自甲基、乙基、苄基。
[0013]
本发明手性γ-甲基苯戊醇的合成方法,包括如下步骤:
[0014]
步骤1:氩气氛围下,将双-(1,5-环辛二烯)镍金属催化剂与手性膦配体在有机溶剂中混合预搅15分钟,随后在氩气保护下,将消旋的炔丙基碳酸酯化合物e、甲烷三甲酸酯化合物f、碱以及路易斯酸加入到混合体系中,在室温至50℃条件下反应12~96小时,薄层色谱点板确定反应终点;随后将该反应体系用乙酸乙酯稀释,并加水萃取,然后将水相用乙酸乙酯萃取3次,合并有机相后用无水硫酸钠干燥并减压浓缩,然后以石油醚/乙酸乙酯(19:1, v/v)作为洗脱剂,通过柱层析分离得到不对称炔丙基化产物g;
[0015]
步骤2:空气环境下,将不对称炔丙基化产物g置于有机溶剂中搅拌,并向反应体系中加入氢氧化钠水溶液(3n),随后将混合物在室温至80℃下搅拌,并通过tlc监测;当反应体系中没有起始原料时,真空除去挥发物,向反应体系中加入乙酸并加热至回流条件下反应 6~12小时;待反应结束后,将乙酸真空浓缩,将该反应体系用乙酸乙酯稀释并加水萃取,然后将水相用乙酸乙酯萃取3次,合并有机相后用无水硫酸钠干燥并减压浓缩,然后以石油醚/乙酸乙酯(1:1,v/v)作为洗脱剂,通过柱层析分离得到化合物h;
[0016]
步骤3:在空气环境下,将化合物h溶解在有机溶剂中,室温下缓慢加入钯/碳,并在一个大气压的氢气氛围下于室温至70℃下反应6~12小时;反应结束后,向反应体系中加入乙酸乙酯稀释,并通过硅藻土过滤,滤液减压浓缩至干,残余物无需纯化即可用于下一步骤;随后在0℃下,向氢化铝锂在有机溶剂的混合物中缓慢加入前一步骤获得的粗产物,使混合物升温至室温下搅拌6~12小时后,在0℃下用十水硫酸钠淬灭反应,并通过硅藻土过滤,滤液减压浓缩至干,然后残余物以石油醚/乙酸乙酯(4:1,v/v)作为洗脱剂,通过柱层析分离得到手性γ-甲基苯戊醇i。
[0017]
步骤1中,双-(1,5-环辛二烯)镍金属催化剂、手性膦配体与甲烷三甲酸酯化合物f的摩尔比为0.1:0.12:1;甲烷三甲酸酯化合物f与消旋的炔丙基碳酸酯化合物e的摩尔当量比为 1:1~3;甲烷三甲酸酯化合物f与碱的摩尔比为1:1~3;路易斯酸的添加量为甲烷三甲酸酯化合物f的0.2倍当量。
[0018]
步骤3中,钯/碳的添加量为化合物h的0.1~0.5倍当量;氢化铝锂的添加量为化合物h 的1.0~2.0倍当量;
[0019]
上述手性γ-甲基苯戊醇的制备过程,可用反应式表示如下:
[0020][0021]
式中:*代表手性碳原子;取代基r
3
选自甲基、乙基、叔丁基;取代基r
4
选自甲基、乙基、苄基。
[0022]
本发明制备过程中,所述碱为碳酸铯、叔丁醇钾、叔丁醇钠、甲醇钠、乙醇钠、双三甲基硅基胺基锂、双三甲基硅基胺基钾或氢化钠。
[0023]
本发明制备过程中,所述有机溶剂为二氯甲烷、乙腈、四氢呋喃、甲苯、甲醇、乙醇、乙酸乙酯、1,2-二氯乙烷、n,n-二甲基甲酰胺或二甲基亚砜。
[0024]
本发明制备过程中,所述手性膦配体为(r或s)-1,1'-联萘-2,2'-双二苯膦、(r或s)-5,5'
-ꢀ
双(二苯基磷酰)-4,4'-二-1,3-联苯、(r或s)-2,2'-双[二(4-甲基苯基膦)]-1,1'-联萘或(r或s)-5,5'
-ꢀ
双(二苯基磷)-四氟-二-1,3-苯二氧杂环。
[0025]
本发明制备过程中,所述路易斯酸为三氟甲磺酸铜、三氟甲磺酸锌、三氟甲磺酸镱或三氟甲磺酸钪。
[0026]
本发明通过不对称炔丙基化反应关键步骤提供了一种合成amg837及手性γ-甲基苯戊醇的方法。本发明以消旋的炔丙基碳酸酯和甲烷三甲酸酯化合物为起始原料,以双-(1,5-环辛二烯)镍金属为催化剂,手性膦试剂为配体,路易斯酸为添加剂,在碱的协助下分别经过不对称炔丙基化反应-水解反应-脱羧反应以及不对称炔丙基化反应-水解反应-脱羧反应-氢化反应-还原反应两条合成路线,均以高收率、高对映选择性及克级规模精准、快速地合成了 amg837及手性γ-甲基苯戊醇化合物。本发明不仅成功开发了amg837及手性γ-甲基苯戊醇的合成方法,很容易制得高产率、高光学纯度的amg837及手性γ-甲基苯戊醇化合物,同时该方法同样具有生物医学实用性和工业应用前景。
附图说明
[0027]
图1是实施例1的制备路线图。
[0028]
图2是实施例2的制备路线图。
具体实施方式
[0029]
实施例1:(-)-amg837的制备
[0030]
1、氮气氛围下,将3-溴苯甲醇(1.0当量)溶解在无水thf中并在冰浴下进行搅拌,随后将三苯膦(1.1当量)分批加入该混合物中,并将偶氮二甲酸二异丙酯(1.1当量)逐滴加入反应体系中。于是将反应混合物在0℃下搅拌1小时,然后在室温下搅拌过夜。反应结束后,将反应混合物在减压条件下进行浓缩,残余物用乙酸乙酯稀释,有机相依次用饱和碳酸氢钠水溶液和氯化钠溶液洗涤,有机相用无水硫酸钠干燥并减压浓缩得到油状残余物。然后以石油醚/乙酸乙酯(9:1)作为洗脱剂,通过柱层析分离得到所需的醚衍生物s1。
[0031]
白色固体(65%)
1
h nmr(400mhz,cdcl3)δ9.89(s,1h),7.80-7.98(m,2h),7.60(s,1h), 7.40
–
7.54(m,1h),7.34
–
3.39(m,1h),7.23
–
7.33(m,1h),7.03
–
7.13(m,2h),5.12(s,2h).
13
c nmr(100mhz,cdcl3)δ190.76,163.34,138.24,132.04,131.41,130.36,130.30,125.85,122.82, 115.11,62.29.
[0032]
2、室温条件下,将甲醇(0.5m),甲苯(0.125m)和碳酸钾水溶液(2.0当量)的溶剂混合物在室温下超声处理30分钟,然后用氮气冲洗30分钟。随后,氮气保护下向反应体系依次加入溴苯衍生物s1(1.0当量),对三氟甲基苯硼酸(1.25当量)和四三苯基膦钯(0.024 当量),将反应混合物在回流条件下搅拌过夜。反应结束后,将反应体系冷却至室温,通过硅藻土过滤不溶物,并将滤液用乙酸乙酯稀释,有机相依次用水,饱和碳酸氢钠水溶液和食盐水洗涤,有机相用无水硫酸钠干燥并减压浓缩得到残余物。然后以石油醚/乙酸乙酯(9:1)作为洗脱剂,通过柱层析分离得到所需的联芳基衍生物s2。
[0033]
白色固体(70%yield)
1
h nmr(400mhz,cdcl3)δ9.92(s,1h),7.88(d,j=8.8hz,2h), 7.66
–
7.82(m,5h),7.48
–
7.70(m,3h),7.12(d,j=8.8hz,2h),5.24(s,2h).
13
c nmr(400mhz, cdcl3)δ190.80,163.57,144.28,140.35,136.78,132.06,130.23,129.42,127.48,127.28,127.22, 126.36,125.80,115.11,70.07.
[0034]
3、向干燥的100ml圆底烧瓶中加入联芳基衍生物s2(10mmol),将烧瓶置于氮气氛围下并冷却至-78℃。随后,向反应体系中缓慢地加入1-丙炔基溴化镁的溶液(22ml,0.5m inthf,1.1当量),并将混合物在-78℃下搅拌1小时,然后使其升温至室温下继续搅拌2小时。待反应结束后,冰浴下将饱和的氯化铵水溶液缓慢地加入到混合物中,水相用乙酸乙酯萃取 3次,合并的有机相用无水硫酸钠干燥并减压浓缩得到炔丙基化合物s3,该化合物无需进一步纯化即可直接用于下一步反应。
[0035]
4、空气环境下,将二碳酸二叔丁酯(1.0当量),三乙胺(1.5当量)和4-二甲氨基吡啶 (1.0当量)溶解在四氢呋喃溶液中(0.3m),然后逐滴加入炔丙基化合物s3(1.0当量),通过tlc监测反应进程,待反应结束后用饱和氯化铵水溶液淬灭反应,并用乙酸乙酯萃取三次,合并的有机相用无水硫酸钠干燥并减压浓缩得到残余物。然后以石油醚/乙酸乙酯(9:1) 作为洗脱剂,通过柱层析分离得到所需的炔丙基碳酸酯原料1。
[0036]
黄色油状(75%yield)
1
h nmr(500mhz,cdcl
3
)δ7.73
–
7.68(m,4h),7.66(s,1h),
7.58
ꢀ–
7.54(m,1h),7.51
–
7.41(m,4h),6.98
–
6.94(m,2h),5.18(q,j=2.2,1h),5.13(s,2h),1.86(d, j=2.1hz,3h),1.31(s,9h).
19
f nmr(470mhz,cdcl
3
)δ-62.40(s).
13
c nmr(125mhz, cdcl
3
)δ158.27,144.58,140.29,138.10,134.70,129.61(q,j=32.1hz),129.40,128.27,127.64, 127.38,127.03,126.49,125.88(q,j=3.8hz),124.41(q,j=270.0hz),114.76,81.91,80.68, 75.41,70.01,64.03,28.61,4.02.atr-ftir(cm-1
):2971,2921,2852,2221,1754,1510,1469, 1367,1326,1240,1126,1072,842,790.esi-ms:calculated[c
29
h
27
f
3
o
4
+na]
+
:519.1754,found: 519.1760.
[0037]
5、在手套箱氮气氛围下,向干燥的100ml schlenk反应管中加入双-(1,5-环辛二烯)镍(132mg,0.48mmol,10mol%)和(r)-5,5'-双(二苯基磷酰)-4,4'-二-1,3-联苯(354mg,0.58 mmol,12mol%),并加入60ml甲苯搅拌15分钟;然后在氮气保护下,将7.2mmol 1(3.58 g,1.5当量),4.8mmol 2(1115mg,1.0当量)和碳酸铯(3.1g,9.6mmol,2.0当量)依次加入到反应管中,并将反应体系在室温下搅拌约96小时,直到底物2完全消耗(通过tlc 监测);随后将该反应体系用乙酸乙酯稀释,并加水萃取,然后将水相用乙酸乙酯萃取3次,合并的有机相用无水硫酸钠干燥并减压浓缩,然后以石油醚/乙酸乙酯(4:1)作为洗脱剂,通过柱层析分离得到不对称炔丙基化产物3(2.65g,90%yield,94%ee)。
[0038]
无色油状(2.65g,90%yield)
1
h nmr(400mhz,cdcl
3
)δ7.73
–
7.67(m,4h),7.66
–
7.62 (m,1h),7.57
–
7.42(m,5h),6.93
–
6.86(m,2h),5.11(s,2h),4.71(q,j=2.3hz,1h),4.24
–ꢀ
4.09(m,6h),1.82(d,j=2.5hz,3h),1.20(t,j=7.1hz,9h).
19
f nmr(375mhz,cdcl
3
)δ
ꢀ-
62.41(s).
13
c nmr(100mhz,cdcl
3
)δ165.45,158.34,144.55,140.27,138.05,131.68,129.62 (q,j=32.0hz),129.38,129.30,127.62,127.34,127.00,126.44,125.86(q,j=4.0hz),124.41(q, j=271.0hz),114.13,80.35,77.35,70.34,69.88,62.08,40.55,13.96,3.87.atr-ftir(cm-1
): 2983,2923,2243,1747,1058,1465,1367,1243,1126,1072,844,790,700.esi-ms:calculated [c
34
h
33
f
3
o
7
+h]
+
:611.2251,found:611.2234.[α]
20d
=-17.3(c=1.00,ch
2
cl
2
).the product was analyzed by hplc to determine the enantiomeric excess:94%ee(ic,hexane/i-proh=95/5, detector:254nm,flow rate:1.0ml/min),t
1
(minor)=19.8min,t
2
(major)=23.8min.
[0039]
6、空气环境下,将不对称炔丙基化产物3(2550mg,4.18mmol)置于40ml甲醇中搅拌,并向反应体系中加入10ml氢氧化钠水溶液(3n),随后将混合物在回流温度下搅拌,并通过tlc监测。当反应体系中没有起始原料时,真空除去挥发物,向反应体系中加入40ml 乙酸并加热至回流条件下反应12小时。待反应结束后,将乙酸真空浓缩,将该反应体系用乙酸乙酯稀释并加水萃取,然后将水相用乙酸乙酯萃取3次,合并有机相后用无水硫酸钠干燥并减压浓缩,然后以石油醚/乙酸乙酯(1:1)作为洗脱剂,通过柱层析分离得到(-)-amg837 (1.62g,88%yield,94%ee)。
[0040]
黄色油状(1.62g,88%yield)
1
h nmr(400mhz,cdcl
3
)δ7.71
–
7.63(m,5h),7.54(d,j= 6.9hz,1h),7.50
–
7.42(m,2h),7.31(d,j=8.3hz,2h),6.95(d,j=8.3hz,2h),5.10(s,2h), 4.10
–
4.01(m,1h),2.87
–
2.64(m,2h),1.82(s,3h).
19
f nmr(375mhz,cdcl
3
)δ-62.34 (s).
13
c nmr(100mhz,cdcl
3
)δ177.25,157.92,144.52,140.26,137.99,133.57,129.60(q,j= 32.0hz),129.40,128.58,127.60,127.38,127.04,126.46,125.86(q,j=3.0hz),124.41(q,j= 271.0hz),115.10,79.50,79.26,70.01,43.42,33.27,3.77.atr-ftir(cm
-
1
):2921,2237,1712, 1508,1328,1243,1124,842,790,700.esi-ms:calculated[c
26
h
21
f
3
o
3
+h]
+
:439.1516found: 439.1524.[α]
20d
=-10.2(c=1.11,ch
2
cl
2
).the product was analyzed by hplc to determine the enantiomeric excess:94%ee(ad-h,hexane/i-proh=80/20,detector:254nm,flow rate:1.0 ml/min),t
1
(major)=12.5min,t
2
(minor)=20.6min.
[0041]
实施例2:(+)-γ-甲基苯戊醇的制备
[0042]
1、在手套箱氮气氛围下,向干燥的100ml schlenk反应管中加入双-(1,5-环辛二烯)镍 (137.5mg,0.5mmol,10mol%)和(r)-5,5'-双(二苯基磷酰)-4,4'-二-1,3-联苯(366.4mg,0.6 mmol,12mol%),并加入60ml二氯甲烷搅拌15分钟;然后在氮气保护下,将7.5mmol 5 (1.85g,1.5当量)(合成参考:org.lett.2015,17,5871.),5.0mmol 2(1160mg,1.0当量),三氟甲磺酸镱(620.2mg,1.0mmol,20mol%)和碳酸铯(3.3g,10.0mmol,2.0当量)依次加入到反应管中,并将反应体系在室温下搅拌约96小时,直到底物2完全消耗(通过tlc监测);随后将该反应体系用乙酸乙酯稀释,并加水萃取,然后将水相用二氯甲烷萃取3次,合并的有机相用无水硫酸钠干燥并减压浓缩,然后以石油醚/乙酸乙酯(19:1)作为洗脱剂,通过柱层析分离得到不对称炔丙基化产物6(1.54g,85%yield,93%ee)。
[0043]
无色油状(1.54g,85%yield)
1
h nmr(500mhz,cdcl
3
)δ7.39
–
7.33(m,2h),7.29
–
7.24 (m,3h),4.29(q,j=7.1hz,6h),3.68(q,j=7.0hz,1h),1.50(d,j=7.0hz,3h),1.29(t,j=7.1 hz,9h).
13
c nmr(125mhz,cdcl
3
)δ165.85,131.67,128.22,127.93,123.53,89.84,82.41, 69.08,62.26,30.88,17.68,14.09.atr-ftir(cm-1
):2981,2923,2854,2242,1743,1598,1463, 1367,1261,1095,862,757,692.esi-ms:calculated[c
20
h
24
o
6
+h]
+
:361.1646,found:361.1650. [α]
20d
=-31.2(c=1.11,ch
2
cl
2
).the product was analyzed by hplc to determine the enantiomeric excess:93%ee(od-h,hexane/i-proh=99/1,detector:254nm,flow rate:1.0 ml/min),t
1
(minor)=11.1min,t
2
(major)=12.8min.
[0044]
2、空气环境下,将不对称炔丙基化产物6(1449mg,4.0mmol)置于40ml甲醇中搅拌,并向反应体系中加入10ml氢氧化钠水溶液(3n),随后将混合物在回流温度下搅拌,并通过tlc监测。当反应体系中没有起始原料时,真空除去挥发物,向反应体系中加入40ml乙酸并加热至回流条件下反应12小时。待反应结束后,将乙酸真空浓缩,将该反应体系用乙酸乙酯稀释并加水萃取,然后将水相用乙酸乙酯萃取3次,合并有机相后用无水硫酸钠干燥并减压浓缩,然后以石油醚/乙酸乙酯(1:1)作为洗脱剂,通过柱层析分离得到化合物7(0.69g, 91%yield)。
[0045]
无色油状(0.69g,91%yield)
1
h nmr(400mhz,cdcl
3
)δ7.42
–
7.35(m,2h),7.28
–
7.26 (m,3h),3.23
–
3.11(m,1h),2.76
–
2.65(m,1h),2.63
–
2.46(m,1h),1.34(d,j=6.9hz,3h).
13
c nmr(125mhz,cdcl
3
)δ177.61 131.77,128.32,127.96,123.53,92.28,81.42,41.50,23.32, 20.87.atr-ftir(cm-1
):2926,2210,1780,1710,1520,1470,846,750.esi-ms:calculated [c
12
h
12
o
2
+h]
+
:189.0910,found:189.0913.
[0046]
3、在空气环境下,将化合物7(685.7mg,3.64mmol)溶解在30ml甲醇溶剂中,室温下缓慢加入10%钯/碳(69mg),并在一个大气压的氢气氛围下于室温下反应12小时;反应结束后,向反应体系中加入乙酸乙酯稀释,并通过硅藻土过滤,滤液减压浓缩至干,残余物无需纯化即可用于下一步骤。随后在0℃下,向氢化铝锂(690.7mg,5.0mmol)在20ml四氢呋喃溶
剂的混合物中缓慢加入前一步骤获得的粗产物,使混合物升温至室温下搅拌12小时后,在0℃下用十水硫酸钠淬灭反应,并通过硅藻土过滤,滤液减压浓缩至干,然后残余物以石油醚/乙酸乙酯(4:1)作为洗脱剂,通过柱层析分离得到(+)-γ-甲基苯戊醇8(0.55g,84% yield,93%ee)。
[0047]
淡黄色油状(0.55g,84%yield)
1
h nmr(400mhz,cdcl
3
)δ7.30
–
7.24(m,2h),7.22
–ꢀ
7.13(m,3h),3.75
–
3.63(m,2h),2.74
–
2.51(m,2h),1.70
–
1.60(m,3h),1.52
–
1.39(m,2h), 1.31(brs,1h),0.97(d,j=6.5hz,3h).
13
c nmr(100mhz,cdcl
3
)δ142.94,128.46,128.44, 125.77,61.20,39.91,39.15,33.48,29.34,19.66.atr-ftir(cm-1
):3344,2929,2869,1604,1496, 1456,746,698.esi-ms:calculated[c
12
h
18
o+h]
+
:179.1430,found:179.1434.[α]
20d
=14.0(c= 0.71,ch
2
cl
2
).the product was analyzed by hplc to determine the enantiomeric excess:93%ee (oj-h,hexane/i-proh=98/2,detector:254nm,flow rate:1.0ml/min),t
1
(minor)=24.9min, t
2
(major)=31.4min.
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips