
 商标分类
商标分类  商标转让
商标转让 
一种甲酰化双环醇及其制备方法与流程
 2021-02-02 10:02:43|
2021-02-02 10:02:43| 376|
376| 起点商标网
起点商标网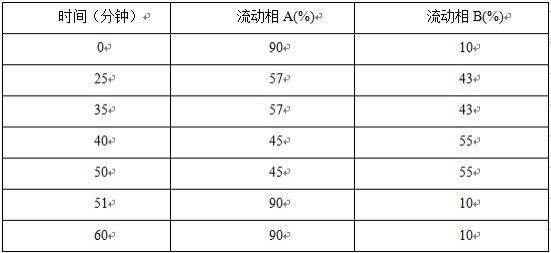
[0001]
本发明属于药物技术领域,具体涉及一种甲酰化双环醇及其制备方法。
背景技术:
[0002]
双环醇是由中国医学科学院、中国协和医科大学药物所开发的产品,是我国第一个拥有自主知识产权的用于治疗慢性病毒性肝炎的国家一类化学新药,用于治疗肝炎所导致的转氨酶升高。在国内,对于双环醇治疗慢性肝炎的临床疗效拥有广泛的应用,动物实验和临床研究表明,双环醇对于肝细胞在多种严重的肝损伤中拥有非常显著的保护作用,可明显改善肝功能、降低转氨酶,同时具有显著的保肝作用和一定的抗病毒效果,停药后不易反弹,安全性好。
[0003]
双环醇为白色或类白色结晶性粉末,无臭、无味,易溶于氯仿和丙酮,溶于乙腈,略溶于乙酸乙酯,微溶于乙醇,不溶于水。其商品名为百赛诺,其化学名为4,4'-二甲氧基-5,6,5'6'-双(亚甲二氧基)-2-羟甲基-2'-甲氧羰基联苯,具有式-结构为:。
[0004]
在双环醇制备过程中和在药品长期性的放置过程中会有杂质生成,而双环醇杂质的存在不仅会影响双环醇的纯度,还可能带来非治疗性的毒副作用。甲酰化双环醇为双环醇制备过程中所生成的双环醇杂质,其结构如下:。
[0005]
目前无对该双环醇杂质甲酰化双环醇及其制备方法的报道,因此对该双环醇杂质甲酰化双环醇及其制备方法和利用合格的杂质标准品进行杂质对照的研究,可用于对双环醇的检验、鉴定以及含量测定,是有效控制双环醇为原料的药物成分和以双环醇为原料的
药物安全性的必要手段。
技术实现要素:
[0006]
本发明提供了一种甲酰化双环醇及其制备方法,可用于生产过程中对双环醇的质量监控的问题。
[0007]
为实现上述发明目的,本发明采用的技术方案如下:一方面,本发明提供了一种甲酰化双环醇,所述甲酰化双环醇的结构式为:。
[0008]
该甲酰化双环醇的化学名称为5'-(((甲酰氧基)甲基)-7,7'-二甲氧基-[4,4'-联苯并[d] [1,3]二恶唑]-5-羧酸甲酯,分子式为c
20
h
18
o
10
,分子量为418.09,本申请发明人在对双环醇的制备工艺探究过程中,意外地发现双环醇粗品中有甲酰化双环醇,由高效液相色谱测定,甲酰化双环醇的含量为0.254%左右,现有技术中没有对甲酰化双环醇的相关报道,也无法从市场上购买得到甲酰化双环醇标准品,该甲酰化双环醇的确定对于有效控制双环醇原料药或其制剂的质量具有重要意义。药品中杂质含量的增加,会影响药品的纯度及其疗效且可能存在较大的安全隐患,将合格的甲酰化双环醇标准品应用于甲酰化双环醇的定性、定量研究和检测,可以为双环醇的安全用药提供保证。
[0009]
另一方面,本发明还提供了一种甲酰化双环醇的制备方法,所述制备方法包括如下步骤:在室温下,向反应溶剂n,n-二甲基甲酰胺中加入双环醇,搅拌后,得到第一溶液;所述第一溶液冷却至第一温度后,滴加三氯氧磷,得到第二溶液;在第二温度下,所述第二溶液发生反应,完全反应后,降温至室温,得到第三溶液;向所述第三溶液中加入水和有机溶剂,萃取,得到有机层;所述有机层浓缩后,所得残留物经柱层析分离纯化,得到目标产物,其中,所述目标化合物为式i结构的甲酰化双环醇。
[0010]
在一个实施例中,所述第一温度为0-10℃。
[0011]
在一个实施例中,所述三氯氧磷和所述双环醇的摩尔比为1.0-3.0。
[0012]
在一个实施例中,所述滴加三氯氧磷过程中,滴加三氯氧磷前后,控制温度不超过10℃。
[0013]
在一个实施例中,所述第二温度为80-100℃。
[0014]
在一个实施例中,所述有机溶剂为乙酸乙酯。
[0015]
在一个实施例中,所述柱层析分离纯化步骤中,使用的洗脱剂为二氯甲烷和石油醚。
[0016]
在一个实施例中,所述二氯甲烷和石油醚的体积比为5:1。
[0017]
本发明提供的甲酰化双环醇及其制备方法,首先通过三氯氧磷先与反应溶剂n,n-二甲基甲酰胺发生反应生成vilsmeier试剂后,vilsmeier试剂再与双环醇发生反应,然后萃取,最后经洗脱剂为二氯甲烷和石油醚的柱层析分离纯化后,得到纯度可达99%以上的目标产物式-结构的甲酰化双环醇;通过定向合成该甲酰化双环醇,可用于对双环醇的检验、鉴定以及含量测定,可达到有效控制以双环醇为原料药物的成分和保证以双环醇为原料药物的安全性的效果,对双环醇原料药物的质量进行有效控制具有重要意义。
附图说明
[0018]
图1为本发明实施例1中甲酰化双环醇的高效液相色谱谱图;图2为本发明实施例1中甲酰化双环醇的高效液相色谱谱图的峰表。
[0019]
图3为本发明实施例1中甲酰化双环醇的质谱谱图;图4为本发明实施例1中甲酰化双环醇的核磁共振氢谱谱图;图5为双环醇粗品与本发明实施例1制得的甲酰化双环醇的高效液相色谱对照谱图;图6为图5中双环醇粗品的高效液相色谱图峰表;图7为双环醇精制品与本发明实施例1制得的甲酰化双环醇的高效液相色谱对照谱图;图8为图7中双环醇精制品的高效液相色谱图峰表。
具体实施方式
[0020]
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0021]
一方面,本发明实施例提供了一种甲酰化双环醇,所述甲酰化双环醇的结构式为:。
[0022]
在双环醇制备过程中会生成式-结构的甲酰化双环醇,该甲酰化双环醇为一种双环醇甲酰化杂质,可通过定向合成该甲酰化双环醇,建立该甲酰化双环醇的检测方法,达到有效控制以双环醇为原料药物的成分和保证以双环醇为原料药物的安全性的效果,对双环醇原料药物的质量进行有效控制具有重要意义。
[0023]
另一方面,本发明还提供了一种甲酰化双环醇的制备方法,所述制备方法包括:步骤s10,在室温下,向反应溶剂n,n-二甲基甲酰胺(dmf)中加入双环醇,搅拌后,得到第一溶液;步骤s20,所述第一溶液冷却至第一温度后,滴加三氯氧磷,得到第二溶液;步骤s30,在第二温度下,所述第二溶液发生反应,完全反应后,降温至室温,得到第三溶液;步骤s40,向所述第三溶液中加入水和有机溶剂,萃取,得到有机层;步骤s50,所述有机层浓缩后,所得残留物经柱层析分离纯化,得到目标产物,其中,所述目标化合物为式-结构的甲酰化双环醇。
[0024]
进一步地,在步骤s20中,在滴加三氯氧磷(pocl
3
)中会大量放热,所以可以采用冰水浴将第一溶液冷却至0-10℃(即所述第一温度为0-10℃)后,再滴加三氯氧磷。
[0025]
其中,在滴加三氯氧磷过程中,放热过大容易导致喷料或爆炸等现象;因此,为了防止大量放热,滴加三氯氧磷前后,控制温度不超过10℃,可采用缓慢滴加等方式来控制。
[0026]
其中,三氯氧磷和双环醇的摩尔比为1.0-3.0,例如可以为1.0、1.5、2.0、2.5、3.0等,当三氯氧磷与双环醇的摩尔比例偏高时,成本增大,当三氯氧磷与双环醇的摩尔比例偏低时,反应不完全。
[0027]
进一步地,在步骤s30中,所述第二温度为80-100℃,例如可以为80℃、81℃、82℃、83℃、84℃、85℃、90℃、95℃、100℃等,当反应的温度过低时,反应不易进行,会导致反应生成的目标产物较少,当反应的温度过高时,有其它副产物生成,会降低反应收率。
[0028]
其中,三氯氧磷先与反应溶剂n,n-二甲基甲酰胺发生反应生成vilsmeier试剂后,vilsmeier试剂再与双环醇发生反应,通过此反应完成了对双环醇的甲酰化过程,得到目标产物甲酰化双环醇。
[0029]
进一步地,在步骤s40中,所述有机溶剂为乙酸乙酯。
[0030]
进一步地,在步骤s50中,所述柱层析分离纯化过程中,使用的洗脱剂为二氯甲烷和石油醚,二氯甲烷和石油醚的体积比为5:1,经洗脱剂为二氯甲烷和石油醚的柱层析分离纯化后,得到的目标产物式-结构的甲酰化双环醇的纯度可达99%以上。
[0031]
按照2015版《中国药典》方法中的的高效液相色谱法对其进行检测,按hplc(高效液相色谱)面积归一法计,确定甲酰化双环醇的纯度,从检测结果可知得到的甲酰化双环醇纯度达99%以上,纯度很高,因此利用该制备方法得到的甲酰化双环醇可以作为标准品使用,可以应用于甲酰化双环醇的定性、定量研究和检测,对有效控制双环醇及其制剂的质量有重要意义。
[0032]
本发明实施例提供的甲酰化双环醇及其制备方法,首先通过三氯氧磷先与反应溶剂n,n-二甲基甲酰胺发生反应生成vilsmeier试剂后,vilsmeier试剂再与双环醇发生反应,然后萃取,最后经洗脱剂为二氯甲烷和石油醚的柱层析分离纯化后,得到纯度可达99%以上的目标产物式-结构的甲酰化双环醇;通过定向合成该甲酰化双环醇,可用于对双环醇
的检验、鉴定以及含量测定,可达到有效控制以双环醇为原料药物的成分和保证以双环醇为原料药物的安全性的效果,对双环醇原料药物的质量进行有效控制具有重要意义。
[0033]
本发明先后进行过多次试验,现举一部分试验结果作为参考,对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
[0034]
实施例1步骤s1:在室温下,将n,n-二甲基甲酰胺(dmf)50ml和双环醇(式-)5.0g(12.8mmol)加入到250ml反应瓶中,搅拌并降温至0-10℃,缓慢滴加三氯氧磷(pocl
3
)1.96g(12.8mmol),滴加完成后,升温至80℃反应2小时,用薄层色谱法(tlc)监测反应,反应完毕后,得到反应液。
[0035]
步骤s2:向上述反应液中加入50ml水和50ml乙酸乙酯,搅拌、静置分层,分出有机层1和水相,水相再用乙酸乙酯提取一次,得到有机层2,合并有机层1和有机层2,得到最终的乙酸乙酯层。
[0036]
步骤s3:在50℃下将上述乙酸乙酯层减压浓缩至干,所得残留物经柱层析分离纯化(洗脱剂为二氯甲烷:石油醚= 5:1),收集含有产物的洗脱液,对洗脱液浓缩后得到白色固体(式-结构的甲酰化双环醇)1.3g。其中,纯度为99.6%,收率为24.1%。
[0037]
反应的合成路线如下:;以下实施例中,反应的合成路线与此实施例相同,便不再赘述。
[0038]
步骤s3得到的白色固体为目标化合物甲酰化双环醇,该目标化合物的纯度检测方式为:以waters xbridge(4.6mm
×
150mm,5μm)为色谱柱,230nm为检测波长,0.01mol/l磷酸二氢钠溶液(调节ph值至6.0)-乙腈=75:25为流动相进行高效液相分析,得甲酰化双环醇的高效液相色谱谱图和峰表分别如图1和图2所示。按高效液相色谱面积归一法计,确定目标产物甲酰化双环醇的纯度为99.63%。
[0039]
目标化合物甲酰化双环醇的质谱谱图如图3所示,检测仪器为waters 3100 mass detector,测试溶剂为甲醇,得到的白色固体的esi-ms(m/z):441 [m+na]
+
,本品分子量为418.09,与目标化合物双环醇杂质分子量相符。
[0040]
目标化合物甲酰化双环醇的核磁共振氢谱谱图如图4所示,核磁共振氢谱谱图表明氢谱数据与目标产物甲酰化双环醇氢相符,说明产物结构正确,具体位移为:
1
hnmr(600mhz,dmso-d
6
)δ:8.092 (s,1h),7.279(s,1h),6.796(s,1h),6.097(d,2h),6.042(d,2h),4.856(q,2h),3.910(s,3h),3.871(s,3h),3.589(s,3h)。
[0041]
实施例2步骤s1:在室温下,将n,n-二甲基甲酰胺50ml和双环醇5.0g(12.8mmol)加入到250ml反
应瓶中,搅拌并降温至0-10℃,缓慢滴加三氯氧磷5.88g(38.3mmol),滴加完成后,升温至100℃反应1小时,tlc监测反应,反应完毕后,得到反应液。
[0042]
步骤s2:向上述反应液中加入50ml水和50ml乙酸乙酯,搅拌、静置分层,分出有机层1和水相,水相再用乙酸乙酯提取一次,得到有机层2,合并有机层1和有机层2,得到最终的乙酸乙酯层。
[0043]
步骤s3:在50℃下将上述乙酸乙酯层减压浓缩至干,所得残留物经柱层析分离纯化(洗脱剂为二氯甲烷:石油醚=5:1),洗脱液浓缩后得到白色固体(式-结构的甲酰化双环醇)1.8g。其中,纯度为99.3%,收率为32.2%。
[0044]
实施例3步骤s1:在室温下,将n,n-二甲基甲酰胺100ml和双环醇10.0g(25.6mmol)加入到500ml反应瓶中,搅拌并降温至0-10℃,缓慢滴加三氯氧磷7.84g(51.3mmol),滴加完后升温至95℃反应1小时,tlc监控原料反应完全后,得到反应液。
[0045]
步骤s2:向上述反应液中加入50ml水和50ml乙酸乙酯,搅拌、静置分层,分出有机层1和水相,水相再用乙酸乙酯提取一次,得到有机层2,合并有机层1和有机层2,得到最终的乙酸乙酯层。
[0046]
步骤s3:在50℃下将上述乙酸乙酯层减压浓缩至干,所得残留物经柱层析分离纯化(洗脱剂为二氯甲烷:石油醚=5:1),洗脱液浓缩后得到白色固体(式-结构的甲酰化双环醇)4.1g。其中,纯度为99.1%,收率为36%。
[0047]
试验例1验证双环醇粗品中是否存在甲酰化双环醇取双环醇粗品,加乙腈-水(体积比为4:1)溶解并稀释制成每1ml中约含1mg的溶液,再精密量取1ml,置200ml量瓶中,用乙腈-水(体积比为4:1)稀释至刻度,摇匀,作为供试品溶液。称取甲酰化双环醇标准品适量,作为对照品溶液。照高效液相色谱法(中国药典2020年版四部通则0512)试验,用十八烷基硅烷键合硅胶为填充剂(welch diamonsil plus c18 4.6mm
ꢀ×
250mm,5
µ
m或效能相当的色谱柱);以0.022%醋酸水溶液(量取纯化水900ml,加入200μl冰乙酸,充分混匀)为流动相a,以乙腈为流动相b,按下表程序进行梯度洗脱;流速为每分钟1.0ml,柱温40℃,检测波长为228nm。精密量取供试品溶液和对照品溶液5
µ
l,注入高效液相色谱仪,记录色谱图,如图5所示,双环醇粗品色谱图对应的峰表如图6所示。
[0048]
图5为双环醇粗品和甲酰化双环醇对照品的高效液相色谱图对照图,1号色谱图为
甲酰化双环醇对照品色谱图,2号色谱图为双环醇粗品色谱图,由图5可知,双环醇粗品中出现保留时间为33.2的甲酰化双环醇的特征峰,与甲酰化双环醇对照品的保留时间一致,因此双环醇粗品中存在甲酰化双环醇。
[0049]
由图6峰表可知,双环醇粗品中甲酰化双环醇的含量为0.257%,远远高于药典2015版第二部中杂质含量不高于0.1%的规定。
[0050]
试验例2验证双环醇精制品中是否存在甲酰化双环醇取双环醇精制品,加乙腈-水(体积比为4:1)溶解并稀释制成每1ml中约含1mg的溶液,再精密量取1ml,置200ml量瓶中,用乙腈-水(体积比为4:1)稀释至刻度,摇匀,作为供试品溶液。称取甲酰化双环醇标准品适量,作为对照品溶液。照高效液相色谱法(中国药典2020年版四部通则0512)试验,用十八烷基硅烷键合硅胶为填充剂(welch diamonsil plus c18 4.6mm
ꢀ×
250mm,5
µ
m或效能相当的色谱柱);以0.022%醋酸水溶液(量取纯化水900ml,加入200μl冰乙酸,充分混匀)为流动相a,以乙腈为流动相b,按下表程序进行梯度洗脱;流速为每分钟1.0ml,柱温40℃,检测波长为228nm。精密量取供试品溶液和对照品溶液5
µ
l,注入高效液相色谱仪,记录色谱图,如图7所示,双环醇精制品色谱图对应的峰表如图8所示。
[0051]
图7为双环醇精制品和甲酰化双环醇对照品的高效液相色谱图对照图,1号色谱图为甲酰化双环醇对照品色谱图,2号色谱图为双环醇精制品色谱图,由图7可知,双环醇精制品中出现保留时间为33.2的甲酰化双环醇的特征峰,与甲酰化双环醇对照品的保留时间一致,因此双环醇精制品中存在甲酰化双环醇。由图8峰表可知,双环醇精制品中甲酰化双环醇的含量为0.067%。
[0052]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips