
 商标分类
商标分类  商标转让
商标转让 
一种内参基因集合及其筛选方法、通用引物组、试剂盒、反应体系及应用与流程
 2021-02-02 09:02:40|
2021-02-02 09:02:40| 389|
389| 起点商标网
起点商标网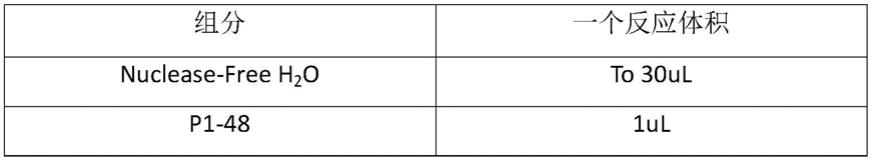
[0001]
本发明属于生物技术领域,尤其涉及一种内参基因集合及其筛选方法、通用引物组、试剂盒、反应体系及应用。
背景技术:
[0002]
第二代高通量测序技术近年来取得了快速发展,和第一代测序技术相比具有速度快、准确率高、成本低等优点。高通量测序技术在新生儿筛查、肿瘤个体化用药、感染性疾病检测、消费基因等领域广泛应用。临床应用过程包括样品前处理、核酸提取、文库构建、上机测序等步骤,为了降低单样品测序成本,一般采用多样本平行处理、核酸提取、文库构建及测序。
[0003]
然而,在多样品平行处理过程中,如在核酸提取时会产生空气与液体表面摩擦的操作步骤均容易产生核酸气溶胶污染,例如,离心机离心、剧烈地摇动反应管或吸样时移液器的反复吸样等操作。在文库构建的pcr过程中,如在接头合成或样品核酸经pcr反应过程中,由于产物的浓度百万倍增加,pcr过程的快速升降温过程也容易产生气溶胶污染。另外,在测序环节中,由于文库内接头的残留,容易产生接头跳跃,从而影响下游数据的分析,产生交叉污染。
[0004]
可见,在二代测序过程中的多个环节易产生交叉污染,从而影响dna检测的准确性,甚至导致假阳性结果。而对于二代测序的阳性内参,目前所用的阳性内参基因对多重pcr反应体系的交叉污染监控效果差强人意。
技术实现要素:
[0005]
本发明实施例提供一种内参基因集合,旨在解决现有的用于多重pcr反应体系的阳性内参基因对反应体系的交叉污染监控效果不好的问题。
[0006]
本发明实施例是这样实现的,一种内参基因集合,所述内参基因集合包括如序列表中的seq id no:1~seq id no:48所示的核苷酸序列或与之相差20个碱基以内的基因中的一个或其任意组合。
[0007]
本发明实施例还提供了上述内参基因集合的筛选方法,包括如下步骤:以斑马鱼的5.7万个基因的cds(编码序列)区作为内参基因备选序列,并过滤gc含量小于40%和大于60%的基因,得到备选内参基因集a;
[0008]
将所述备选内参基因集a的基因序列按照等比切分成3份,分别统计gc含量,并过滤掉任何一段的gc含量小于40%和大于60%的基因以及gc集中的基因,得到备选内参基因集b;
[0009]
过滤所述备选内参基因b中的含连续5个相同碱基的基因,得到所述内参基因集合。
[0010]
本发明实施例还提供了一种应用于多重pcr体系的通用引物组,所述通用引物组
包括:用于连接所述内参基因两端特异性核酸片段的通用接头,以及分别与如上所述的内参基因集合的每个内参基因对应的引物对或者与所述引物对相差一个碱基的dna序列;其中每对引物对均由一正向引物和一反向引物组成,每对所述引物对的正向引物和反向引物的核苷酸序列依次分别为序列表中的seq id no:49~seq id no:144。
[0011]
本发明实施例还提供了一种应用于多重pcr体系的试剂盒,所述试剂盒包括如上所述的内参基因集合以及如上所述的应用于多重pcr体系的通用引物组。
[0012]
本发明实施例还提供了应用于多重pcr体系的反应体系,所述反应体系含有如上所述的内参基因集合以及如上所述的应用于多重pcr体系的通用引物组。
[0013]
本发明实施例还提供了如上所述的内参基因集合在维生素代谢基因检测中的应用。
[0014]
本发明实施例提供的内参基因集合中的内参基因适用于illumina(二代测序)、thermo fisher scientific的测序平台,可应用于多重pcr体系的核酸提取及第二代高通量测序等环节的阳性对照和交叉污染监控,并且还可应用于维生素代谢基因的检测,阳性对照和交叉污染监控的效果好。
具体实施方式
[0015]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合具体实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0016]
本发明实施例提供的内参基因集合适用于illumina(二代测序)、thermo fisher scientific的测序平台,可应用于多重pcr体系的核酸提取及第二代高通量测序等环节的阳性对照和交叉污染监控,且阳性对照和交叉污染监控的效果好。
[0017]
本发明实施例提供了一种内参基因集合,该内参基因集合中的内参基因来源于斑马鱼基因loc100148225、zp3、loc100536860、bdkrb2、crls1、loc568955、shprh、mapkbp1、hnrnpua、fbxo33、samd4a、kcnk13a、ryr2b、hmx2、ttbk1b、exo1、birc6、wdpcp、morn2、wapla、jade1、sptbn5、tecpr2、adam17a、ralgapa2、plk4、myt1lb、tgfb3、asb2b、loc100535674、usp8、tm2d3、eif2a、spata5l1、cyp1a、grm3、bicd1a、hipk2、vps9d1、igf1ra、bcar1、gnrhr4、znf710a、malt3、syt9a、loc100535882、b3gat1a、gyltl1b。
[0018]
5等基因的部分序列。上述斑马鱼参考基因组可从ncbi(national center for biotechnology information,是美国国立生物技术信息中心,美国国立卫生研究院(nih)分支机构美国国家医学图书馆(nlm)的一部分)数据库获得。
[0019]
所述内参基因集合包括如序列表中的seq id no:1~seq id no:48所示的核苷酸序列或与之相差20个碱基以内的基因中的一个或其任意组合。
[0020]
在本发明实施例中,所述内参基因集合中的每个内参基因的基因长度为180~250bp(碱基对),gc含量为40%~60%,碱基g、c不集中在dna的3
’
或5
’
端,连续碱基为5个以内。gc含量是指在dna的4种碱基中,鸟嘌呤和胞嘧啶所占的比率称为gc含量。在双链dna中,腺嘌呤与胸腺嘧啶(a/t)之比,以及鸟嘌呤与胞嘧啶(g/c)之比都是1。但是,(a+t)/(g+c)之比则随dna的种类不同而异。gc含量愈高,dna的密度也愈高,同时热及碱不易使之变性,因此利用这一特性便可进行dna的分离或测定。
[0021]
本发明实施例还提供了上述内参基因集合的筛选方法,包括如下步骤:
[0022]
以斑马鱼的5.7万个基因的cds(编码序列)区作为内参基因备选序列,并过滤gc含量小于40%和大于60%的基因,得到备选内参基因集a。
[0023]
将所述备选内参基因集a的基因序列按照等比切分成3份,分别统计gc含量,并过滤掉任何一段的gc含量小于40%和大于60%的基因以及gc集中的基因,得到备选内参基因集b。
[0024]
过滤所述备选内参基因b中的含连续5个相同碱基的基因,得到所述内参基因集合。
[0025]
本发明实施例还提供了一种应用于多重pcr体系的通用引物组,所述通用引物组包括:用于连接所述内参基因两端特异性核酸片段的通用接头,以及分别与如权利要求1所述的内参基因集合的每个内参基因对应的引物对或者与所述引物对相差一个碱基的dna序列;其中每对引物对均由一正向引物和一反向引物组成,每对所述引物对的正向引物和反向引物的核苷酸序列依次分别为序列表中的seq id no:49~seq id no:144。
[0026]
其中,上述通用接头包括第一通用接头和第二通用接头;所述第一通用接头的核苷酸序列为:tacacgacgctcttccgatct;所述第二通用接头的核苷酸序列为:tccttggcacccgagaattcca。
[0027]
其中,上游引物5
’-
3
’
依次为第一通用接头和内参基因特异性上游引物序列;所属下游引物5
’-
3
’
依次为第二通用接头和内参基因特异性下游引物序列。
[0028]
本发明实施例还提供了一种应用于多重pcr体系的试剂盒所述试剂盒包括如上所述的内参基因集合以及如上所述的应用于多重pcr体系的通用引物组。
[0029]
本发明实施例还提供了一种应用于多重pcr体系的反应体系,所述反应体系含有如上所述的内参基因集合以及如上所述的应用于多重pcr体系的通用引物组。
[0030]
本发明实施例提供的上述内参基因集合可应用于维生素代谢基因检测,以作为阳性对照和交叉污染的监控内参。
[0031]
其应用方法如下:
[0032]
获取n个待测核酸样本;向每份待测样本中加入1000~3000个拷贝的内参基因;提取每份待测核酸样本的总dna,放置4℃保存,备用(待进入多重pcr建库环节时使用);将上述提取好的n个待检测核酸样本进行两轮pcr扩增,构建测序dna文库;采用illumina或thermo fisher scientific的测序平台对上述的pcr扩增产物进行测序。
[0033]
下面结合具体的实施例对本发明的技术效果做进一步的描述。其中实验中所用到试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品,例如,可以采购自illumina、天根生化公司等。
[0034]
实施例1内参基因集合的筛选方法
[0035]
常见模式生物有斑马鱼、拟南芥、线虫、果蝇、小鼠,模式生物具有基因组信息研究透彻和易获得性,相比人工模拟随机生成人工合成序列具有成本低等特点。本发明选择斑马鱼作为内参基因集信息来源。从ncbi获得斑马鱼参考基因组,斑马鱼一共包含57k个基因。
[0036]
筛选步骤如下:
[0037]
(1)基于斑马鱼57k个基因的cds(编码序列)区作为内参基因备选序列,评估gc含
量,过滤gc含量小于40%和大于60%的基因,得到备选内参基因集a。
[0038]
(2)将内参基因集a的基因序列按照等比切分成3份,分别统计gc含量,过滤任何一段的gc含量小于40%和大于60%的基因,过滤gc集中的基因,得到备选内参基因集b。
[0039]
(3)过滤所述备选内参基因b中的含连续5个相同碱基的基因,得到内参基因集合c,即本发明的内参基因集合。
[0040]
(4)采用primer3 plus引物设计软件对内参基因集c筛选目标引物,扩增子长度180-250bp。
[0041]
斑马鱼基因集合经过上述条件过滤,得到一组包含48个内参基因及其多重pcr体系通用引物组,内参基因序列如序列表中的seq id no:1~seq id no:48所示,多重pcr体系通用引物如序列表中的seq id no:48~seq id no:144所示。
[0042]
实施例2内参基因集合的筛选及其对应引物的验证
[0043]
以上述实施例1筛选获得的内参基因集的48个内参基因为模板,设计并合成与该内参基因集中的每个内参基因对应的多重pcr体系引物对,将引物稀释到100um(毫摩尔/升),等体积(ul)混合引物得到内参基因mix(混合物),按照以下流程进行测序文库构建,验证内参基因集和引物对敏感性和扩增效率差异。
[0044]
基于多重pcr技术的第二代高通量测序文库构建及纯化:
[0045]
(1)对以上获得的内参基因引物p1 mix,进行两轮pcr扩增的测序文库构建;按如下表1所示的体系,配置第一轮pcr扩增体系,斑马鱼基因组模板(50ng)、p1 mix、3x enzyme、ddh
2
o混合,得预混好多的重pcr反应液mix。
[0046]
表1
[0047]
组分一个反应体积nuclease-free h
2
oto 30ulp1 mix8ul斑马鱼gdna(50ng)10ul3x enzyme10ul
[0048]
(2)将上述预混好多的重pcr反应液mix放置于bio-rad t100(美国bio-rad伯乐t100型梯度pcr仪),进行按以下参数进行第一轮pcr扩增,程序如下:先98℃,3min,然后98℃,20秒,60℃,6min进行17~20个循环,然后72℃,2min,最后10℃直至收取pcr产物。
[0049]
(3)然后将上述经过扩增的pcr反应液加入0.5倍体积的ampure xp beads,使用移液器上下吹打,以使扩增产物与ampure xp beads充分混匀。室温静置3分钟。
[0050]
(4)将上述经过静置处理的混合物置于磁力架上,静置3min,待磁珠完全被吸附后,用移液器吸取上清至新的ep管/96孔板中,保留上清液(避免吸到磁珠)。
[0051]
(5)将上述保留上清液中加入0.6倍pcr体积ampurexp beads,使用移液器上下吹打,以使扩增产物与ampure xp beads充分混匀。室温静置3分钟。
[0052]
(6)将上述经过静置处理的合物置于磁力架上,静置3min,待磁珠完全被吸附后,溶液完全澄清,用移液器小心移除上清,保留磁珠。
[0053]
(7)将上述保留的磁珠加入50ul磁珠洗液1,悬浮磁珠,室温静置2分钟。用强力磁铁或磁力架吸附磁珠,直至溶液澄清为止。用移液器吸弃上清,留磁珠。
[0054]
(8)将上述所得磁珠加入100ul 80%乙醇,用磁力架反复在不同两面来回吸附磁
珠以充分悬浮磁珠以洗涤。
[0055]
(9)将上述所得充分重悬的第四混合液置于磁力架上,室温静置3分钟,直至溶液澄清为止。用移液器小心去除上清,避免吸到磁珠。
[0056]
(10)将上述所得的磁珠,室温放置5分钟,直至乙醇挥发干净。本步骤亦可放于50℃烘箱3~5分钟左右。
[0057]
(11)将上述所得到的磁珠,按照如下表2的体系配置第二轮pcr扩增体系(p2引物对为带标签信息的通用引物,序列分别为:
[0058]
正向引物:aatgatacggcgaccaccgagatctacactctttccctacacgacgctcttccgatct;
[0059]
反向引物:caagcagaagacggcatacgagatgtgactgga gttccttggcacccgagaattcca)。
[0060]
表2
[0061]
组分一次反应体积nuclease-free h
2
o18ul3x enzyme ht10ulp22ul总体积30ul
[0062]
(12)将上述预混好的pcr反应液放置bio-rad t100,按以下参数进行第二轮pcr扩增,程序如下:先98℃、2min,然后98℃、15sec,60℃、15sec,72℃、30sec进行6~9个循环,然后72℃,2min,最后10℃直至收取pcr产物。
[0063]
(13)将上述经过梯度扩增的pcr反应液加入0.8倍体积的ampure xp beads,使用移液器上下吹打,以使pcr产物与ampure xp beads充分混匀。室温静置3分钟。
[0064]
(14)将上述得到的混合液置于磁力架吸附磁珠,静置2分钟,直至溶液澄清为止。用移液器小心吸弃上清,保留磁珠。
[0065]
(15)将上述得到的磁珠加入100ul 80%乙醇,使用磁力架反复在不同的两面来回吸附磁珠以充分悬浮磁珠以洗涤,使用移液器小心吸弃上清,保留磁珠。
[0066]
(16)将上述得到的磁珠室温放置5分钟,直至乙醇挥发干净。本步骤亦可放于50℃烘箱3-5分钟左右。
[0067]
(17)将上述得到的磁珠加入20ul elution buffer(洗脱缓冲液),充分悬浮磁珠,得到混合液七,室温静置2min以洗脱dna。
[0068]
(18)将上述磁珠置于磁力架静置5分钟,直至溶液完全澄清为止,将上清吸至一新的1.5/0.5/0.2ml离心管或96孔pcr管。
[0069]
将上述所得文库送至北京诺禾致源科技股份有限公司采用illumin novaseq测序平台进行测序,测序结果数据质控结果如下表3所示:
[0070]
表3
[0071]
样本名称total readsclean readsadapter rateq20q30gc含量icg-4810,221,5369,752,2342.7992.0387.4652.2%
[0072]
统计48个备选内参基因reads数和相对丰度如下表4所示:表4中的引物id p_ic01对应的是通用引物组seq id no:49和seq id no:50的测试结果,引物id p_ic02对应的是通用引物组seq id no:51和seq id no:52的测试结果.....以此类推,引物id p_ic48对应的是通用引物组seq id no:143和seq id no:144的测试结果。
[0073]
表4
[0074]
引物id序列数引物id序列数引物id序列数引物id序列数p_ic01120619p_ic1323664p_ic25187443p_ic3716977p_ic0238684p_ic1469077p_ic2656586p_ic38195408p_ic03119407p_ic15120925p_ic2762959p_ic39151803p_ic04157015p_ic1628918p_ic28162066p_ic40125056p_ic0583431p_ic17177046p_ic298287p_ic41118585p_ic0648447p_ic18194287p_ic30167505p_ic42198621p_ic0785737p_ic19123271p_ic31107430p_ic4338928p_ic08113956p_ic2091308p_ic3213772p_ic44130023p_ic0915300p_ic2118034p_ic33138889p_ic45151942p_ic1096976p_ic2298643p_ic3419432p_ic4678105p_ic119835p_ic23183223p_ic35199866p_ic47138683p_ic12198795p_ic24170039p_ic36104481p_ic4898561
[0075]
从上表4的测试结果可以看出,本发明实施例的内参基因在多重pcr体系中均为阳性。通过分析48个内参基因的扩增效率f(扩增子reads数/所有扩增子reads中位数)可得,扩增效率f中位值1.07(q1=0.45、q3=1.45),符合生产应用。
[0076]
实施例3内参基因集合的制备
[0077]
选择上述实施例2通过验证的内参基因集合中的48个内参基因为模板,合成内参基因特异性引物对,将引物稀释到10um,按照如下表5的体系配置pcr反应体系:斑马鱼基因组模板(50ng)、特异性引物p1-p48(即与上述48个内参基因对应的48组通用引物)、pcr预混液、ddh
2
o混合。
[0078]
表5
[0079][0080][0081]
将上述预混好多的重pcr反应液mix放置于bio-rad t100,进行按以下参数进行第一轮pcr扩增,程序如下:先94℃、2min,然后98℃、10s,60℃、30s、68℃、30s进行35个循环,然后68℃、5min,最后4℃长期。
[0082]
将上述扩增得到的pcr产物用美基生物pcr pure lq kit试剂盒进行纯化,之后采用qubit4.0测定核酸浓度,并根据浓度到拷贝数的换算公式换算得核酸片段拷贝数,得到每个内参基因浓度,稀释内参基因,将内参基因浓度稀释到1000~1500copies/ul浓度,得到最终内参基因集合,做好标签备用。
[0083]
换算方法如下:
[0084]
1)从copies/μl到ng/μl:
[0085]
首先已知样本中核酸的大小和每个拷贝dna的ng值,以人基因组dna为例:将dna大小换算成ng/copy:人基因组dna大小(mb):3000mb/copy x 1bp的分子量:1.096x10-21g/bp x将mb转换成bp:1x106bp/mb x将g转换成ng:1x109ng/g=每个拷贝dna的ng值:0.0033ng/copy。然后只需将拷贝数浓度直接换算即可,如果拷贝数浓度是3000copies/μl,那么如下公式转换:3000copies/μl x 3.3pg/copy(0.0033ng/copy)=9.9ng/μl。
[0086]
2)从ng/ul换算到copies/ul:
[0087]
首先已知样本中核酸的大小,以人基因组大小为例是3000mb/copy,换算到每个copy的核酸分子的分子量ng。人基因组dna大小(mb):3000mb/copyx 1bp的分子量:1.096x10-21g/bp x将mb转换成bp1x106bp/mb x将g转换成ng:1x10-9ng/g=每个拷贝dna的ng值:0.0033ng/copy。如质量浓度为10ng/ul,按照如下进行计算:10ng/ul=3030copies/ul;0.0033ng/copy=3030copies/ul。
[0088]
实施例4内参基因集合在维生素代谢基因检测中的应用
[0089]
(1)内参投入与核酸提取
[0090]
选取10例体积为2ml的唾液样品,样本前处理过程中,加入5ul内参基因,做好标记;采用天根tianamp swab dna kit(dp322-01)试剂盒进行核酸提取,对提取的基因组采用nanodrop 2000进行纯度检测,采用qubit4.0测定浓度,都大于0.2ng/ul。
[0091]
(2)基于多重pcr方法的dna标签文库构建
[0092]
对待测10例唾液核酸样本,采用人体密码基因科技的维生素代谢基因检测panel进行文库构建,结合维生素代谢基因检测方法(具体方法参照上述实施例2),进行文库构建,经过第一轮特异性引物对扩增后,第二轮pcr不同样本的反应体系添加不同的标签引物,对pcr产物纯化后得到待上机文库。
[0093]
(3)测序与数据分析
[0094]
对以上10个纯化后dna文库样品按照等摩尔数混合为文库mix,经agilent2100检测合格后,采用illumina novaseq平台进行测序,测序前准备及测序详细步骤参见novaseq reagent kits操作说明书。
[0095]
测序下机数据进行标签识别,用于区分不同样本的测序数据集;通过对下机的测序得到的序列进行比对、过滤、去重等质控处理,得到内参基因检出序列数,结果如表6所示;样本目标区域靶向捕获的有效覆盖深度进行统计及单碱基突变分析。结果显示,本发明实施提供的内参基因在维生素代谢基因检测过程中起到阳性对照、交叉污染监控的作用。
[0096]
表6 10个唾液样本内参基因需列数分布情况
[0097]
样品编号total_readsclean_reads内参基因内参基因序列数内参基因(污染)s01402000373860p_ic012568nas02386116342088p_ic021587nas03315439293582p_ic03305p_ic01(3条)s04443728418367p_ic048091nas05441320411588p_ic054307p_ic08(12条)s06344375321067p_ic06724nas07413914314940p_ic07578nas08428827396930p_ic089862na
s09382319352557p_ic095278nas10434126403452p_ic103834na
[0098]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips