
 商标分类
商标分类  商标转让
商标转让 
一种苯并菲衍生物及其应用的制作方法
 2021-02-02 06:02:42|
2021-02-02 06:02:42| 276|
276| 起点商标网
起点商标网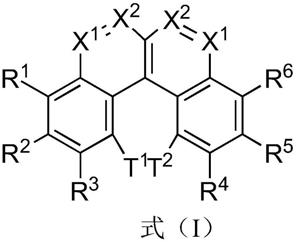
[0001]
本发明属于有机电致发光技术领域,具体涉及一种苯并菲衍生物及其应用。
背景技术:
[0002]
有机电致发光元件所使用的物质大部分为纯有机物或有机物与金属形成络合物的有机金属络合物,根据用途可区分为空穴注入物、空穴输送物、发光物、电子输送物、电子注入物等。在此,作为空穴注入物或空穴输送物主要使用离子化能相对较小的有机物,作为电子注入物或电子输送物主要使用电负性较大的有机物。此外,作为发光辅助层所使用的物质最好能满足如下特性。
[0003]
第一、有机电致发光元件中所使用的物质需较好的热稳定性,其原因是在有机电致发光元件内部因电荷的迁移而发生焦耳热,目前,作为空穴输送层通常所使用的材料的玻璃化温度低,因此在低温下驱动时出现因发生结晶化而引起发光效率降低的现象。第二、为了降低驱动电压,需要与阴极和阳极邻接的有机物设计成电荷注入势垒较小,电荷迁移率高。第三、电极和有机层的界面、有机层和有机层的界面上一直存在能量壁垒而不可避免地累积一些电荷,因此需要使用电化学稳定性优异的物质。
[0004]
发光层由主发光体和掺杂物这两种物质构成,掺杂物需要量子效率高,与掺杂物相比主发光体需要能隙大而容易发生向掺杂物的能量转移。用于电视、移动设备等的显示器根据红色、绿色、蓝色这三原色实现全彩色,发光层分别由红色主发光体/掺杂物、绿色主发光体/掺杂物以及蓝色主发光体/掺杂物构成。目前蓝光材料仍然存在发光量子效率低、色纯度差的问题。造成这种状况的主要原因是因为蓝光来自于能隙较宽的能级间的跃迁,而宽禁带的有机化合物在进行分子设计时存在一定的困难,其次蓝光材料的体系中存在着较强的π-π键相互作用,有着很强的电荷转移特性,从而使宽带隙中存在更多的无辐射弛豫通道,加剧了分子之间荧光淬灭,降低了蓝光体系的量子产率。因此设计合成综合性能优良的蓝光材料,将成为有机电致发光材料研究的重要课题。
[0005]
鉴于以上原因,特提出本发明。
技术实现要素:
[0006]
为了解决现有技术存在的以上问题,本发明提供了一种苯并菲衍生物及其应用,本发明的所述的苯并菲衍生物应用与有机电致发光材料中,可以提高有机电致发光元件的效率、寿命。
[0007]
本发明的第一目的,提供了一种苯并菲衍生物,所述的苯并菲衍生物的结构式如式(i)所示:
[0008][0009]
其中,r1、r2、r3、r4、r5、r6、t1、t2各自独立地选自氢、氘、具有c
1-c
40
的直链烷基、具有c
1-c
40
的直链杂烷基、具有c
3-c
40
的支链或环状的烷基、具有c
3-c
40
的支链或环状的杂烷基、具有c
2-c
40
的烯基或炔基、具有5~60个原子的芳族环系或杂芳族环系中的一种,r1、r2、r3、r4、r5、r6、t1、t2中的每个基团可被一个或多个基团r取代;
[0010]
优选的,t1和t2通过选自o、s、s(=o)、s(=o)2、nr、c(r)2、br、pr、p(=o)r、si(r)2的桥连基彼此桥连;
[0011]
x1、x2如式(ii)所示:
[0012][0013]
其中,g为c(r)2、nr、氧或硫,z在每次出现时相同或不同,选自cr或n,并且^指示式(i)中的相邻基团x1、x2;
[0014]
所述r在每次出现时相同或不同,选自氢原子、氘原子、卤素原子、腈基、硝基、n(ar1)2、n(r7)2、c(=o)ar1、c(=o)r7、p(=o)(ar1)2、具有c
1-c
40
的直链烷基、具有c
1-c
40
的直链杂烷基、具有c
3-c
40
的支链或环状的烷基、具有c
3-c
40
的支链或环状的杂烷基、具有c
2-c
40
的烯基或炔基、具有5至80个、优选5至60个原子的芳族环或杂芳族环系、具有5至60个原子的芳氧基或杂芳氧基中的一种,r中的每个基团可被一个或多个基团r7取代,或这些体系的组合,其中一个或多个非相邻的-ch
2-基团可被r7c=cr7、c≡c、si(r7)2、ge(r7)2、sn(r7)2、c=o、c=s、c=se、c=nr7、p(=o)(r7)、so、so2、nr7、o、s或conr7代替,并且其中一个或多个氢原子被氘原子、卤素原子、腈基或硝基代替,其中两个或更多个相邻的r可以任选地接合或稠合形成单环或多环的脂族、芳族或杂芳族环系,所述环系可被一个或多个基团r7取代;
[0015]
r7在每次出现时相同或不同,选自氢原子、氘原子、卤素原子、腈基、硝基、n(ar1)2、n(r8)2、c(=o)ar1、c(=o)r8、p(=o)(ar1)2、具有c
1-c
40
的直链烷基、具有c
1-c
40
的直链杂烷基、具有c
3-c
40
的支链或环状的烷基、具有c
3-c
40
的支链或环状的杂烷基、具有c
2-c
40
的烯基或炔基、具有5至60个原子的芳族环或杂芳族环系、具有5至60个原子的芳氧基或杂芳氧基中的一种,r7中的每个基团可被一个或多个基团r8取代,或这些体系的组合,其中一个或多
甲基丁氧基、正己氧基、环己氧基、正庚氧基、环庚氧基、正辛氧基、环辛氧基、2-乙基己氧基、五氟乙氧基和2,2,2-三氟乙氧基。杂烷基优选具有1-40个碳原子的烷基,是指其中单独的氢原子或-ch
2-基团可被氧、硫、卤素原子取代的基团,被认为是指烷氧基、烷硫基、氟代的烷氧基、氟代的烷硫基,特别是指甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、仲丁氧基、叔丁氧基、甲硫基、乙硫基、正丙硫基、异丙硫基、正丁硫基、异丁硫基、仲丁硫基、叔丁硫基、三氟甲硫基、三氟甲氧基、五氟乙氧基、五氟乙硫基、2,2,2-三氟乙氧基、2,2,2-三氟乙硫基、乙烯氧基、乙烯硫基、丙烯氧基、丙烯硫基、丁烯硫基、丁烯氧基、戊烯氧基、戊烯硫基、环戊烯氧基、环戊烯硫基、己烯氧基、己烯硫基、环己烯氧基、环己烯硫基、乙炔氧基、乙炔硫基、丙炔氧基、丙炔硫基、丁炔氧基、丁炔硫基、戊炔氧基、戊炔硫基、己炔氧基、己炔硫基。
[0025]
一般来说,根据本发明的环烷基、环烯基可为环丙基、环丁基、环戊基、环己基、环丁烯基、环戊烯基、环己烯基、环庚基、环庚烯基,其中一个或多个-ch2-基团可被上述基团代替;此外,一个或多个氢原子还可被氘原子、卤素原子或腈基代替。
[0026]
根据本发明的芳族或杂芳族环原子,在每种情况下还可被上述基团r8取代的芳族或杂芳族环系,特别是指衍生自如下物质的基团:苯、萘、蒽、苯并蒽、菲、芘、苝、荧蒽、并四苯、并五苯、苯并芘、联苯、偶苯、三联苯、三聚苯、芴、螺二芴、二氢菲、二氢芘、四氢芘、顺式或反式茚并芴、顺式或反式茚并咔唑、顺式或反式吲哚并咔唑、三聚茚、异三聚茚、螺三聚茚、螺异三聚茚、呋喃、苯并呋喃、异苯并呋喃、二苯并呋喃、噻吩、苯并噻吩、异苯并噻吩、二苯并噻吩、吡咯、吲哚、异吲哚、咔唑、吡啶、喹啉、异喹啉、吖啶、菲啶、苯并[5,6]喹啉、苯并[6,7]喹啉、苯并[7,8]喹啉、吩噻嗪、吩噁嗪、吡唑、吲唑、咪唑、苯并咪唑、萘并咪唑、菲并咪唑、吡啶并咪唑、吡嗪并咪唑、喹喔啉并咪唑、噁唑、苯并噁唑、萘并噁唑、蒽并噁唑、菲并噁唑、异噁唑、1,2-噻唑、1,3-噻唑、苯并噻唑、哒嗪、六氮杂苯并菲、苯并哒嗪、嘧啶、苯并嘧啶、喹喔啉、1,5-二氮杂蒽、2,7-二氮杂芘、2,3-二氮杂芘、1,6-二氮杂芘、1,8-二氮杂芘、4,5-二氮杂芘,4,5,9,10-四氮杂苝、吡嗪、吩嗪、吩噁嗪、吩噻嗪、荧红环、萘啶、氮杂咔唑、苯并咔啉、咔啉、菲咯啉、1,2,3-三唑、1,2,4-三唑、苯并三唑、1,2,3-噁二唑、1,2,4-噁二唑、1,2,5-噁二唑、1,3,4-噁二唑、1,2,3-噻二唑、1,2,4-噻二唑、1,2,5-噻二唑、1,3,4-噻二唑、1,3,5-三嗪、1,2,4-三嗪、1,2,3-三嗪、四唑、1,2,4,5-四嗪、1,2,3,4-四嗪、1,2,3,5-四嗪、嘌呤、蝶啶、吲嗪和苯并噻二唑或者衍生自这些体系的组合的基团。
[0027]
进一步地,所述的苯并菲衍生物的结构主要包括cjhb271~cjhb480,其中g选自氧、硫、ch2、c(ch3)2、c(c6h5)2、n-ch3、n-c2h5、n-c6h5中的一种:
[0028]
[0029]
[0030]
[0031]
[0032]
[0033]
[0034]
[0035]
[0036]
[0037]
[0038]
[0039][0040]
本发明的第二目的,提供了一种所述的苯并菲衍生物在有机电致发光材料中的应用。
[0041]
进一步地,所述的有机电致发光材料为有机电致发光元件用材料、有机场效应晶体管用材料或有机薄膜太阳电池用材料。
[0042]
有机电致发光材料可以单独使用本发明的苯并菲衍生物构成,也可以同时含有其他化合物。
[0043]
本发明的第三目的,提供了一种有机电致发光装置,所述的有机电致发光装置包括第一电极、第二电极和置于所述第一电极和第二电极之间的至少一层有机层,至少一层有机层包含所述的苯并菲衍生物。
[0044]
所述有机电致发光装置包含阴极、阳极和至少一个发光层。除了这些层之外,它还可以包含其它的层,例如在每种情况下,包含一个或多个空穴注入层、空穴传输层、空穴阻挡层、电子传输层、电子注入层、激子阻挡层、电子阻挡层和/或电荷产生层。具有例如激子阻挡功能的中间层同样可引入两个发光层之间。然而,应当指出,这些层中的每个并非必须都存在。此处所述有机电致发光装置可包含一个发光层,或者它可包含多个发光层。即,将能够发光的多种发光化合物用于所述发光层中。特别优选具有三个发光层的体系,其中所述三个层可显示蓝色、绿色和红色发光。如果存在多于一个的发光层,则根据本发明,这些层中的至少一个层包含本发明的化合物。
[0045]
进一步地,根据本发明的有机电致发光装置不包含单独的空穴注入层和/或空穴传输层和/或空穴阻挡层和/或电子传输层,即发光层与空穴注入层或阳极直接相邻,和/或发光层与电子传输层或电子注入层或阴极直接相邻。
[0046]
在根据本发明的有机电致发光装置的其它层中,特别是在空穴注入和空穴传输层中以及在电子注入和电子传输层中,所有材料可以按照根据现有技术通常所使用的方式来使用。本领域普通技术人员因此将能够在不付出创造性劳动的情况下与根据本发明的发光层组合使用关于有机电致发光元件所知的所有材料。
[0047]
此外优选如下的有机电致发光装置,其特征在于借助于升华方法施加一个或多个层,其中在真空升华装置中在低于10-5
pa、优选低于10-6
pa的初压下通过气相沉积来施加所述材料。然而,所述初压还可能甚至更低,例如低于10-7
pa。
[0048]
同样优选如下的有机电致发光器件,其特征在于借助于有机气相沉积方法或借助
于载气升华来施加一个或多个层,其中,在10-5
pa至1pa之间的压力下施加所述材料。该方法的特别的例子是有机蒸气喷印方法,其中所述材料通过喷嘴直接施加,并且因此是结构化的。
[0049]
此外优选如下的有机电致发光装置,从溶液中,例如通过旋涂,或借助于任何所希望的印刷方法例如丝网印刷、柔性版印刷、平版印刷、光引发热成像、热转印、喷墨印刷或喷嘴印刷,来产生一个或多个层。可溶性化合物,例如通过适当的取代获得可溶性化合物。这些方法也特别适于低聚物、树枝状大分子和聚合物。此外可行的是混合方法,其中例如从溶液中施加一个或多个层并且通过气相沉积施加一个或多个另外的层。
[0050]
这些方法是本领域普通技术人员通常已知的,并且他们可以在不付出创造性劳动的情况下将其应用于包含根据本发明的化合物的有机电致发光元件。
[0051]
因此,本发明还涉及制造根据本发明的有机电致发光装置的方法,其特征在于借助于升华方法来施加至少一个层,和/或特征在于借助于有机气相沉积方法或借助于载气升华来施加至少一个层,和/或特征在于从溶液中通过旋涂或借助于印刷方法来施加至少一个层。
[0052]
此外,本发明涉及包含至少一种上文指出的本发明的稠环芳香族化合物。如上文关于有机电致发光装置指出的相同优选情况适用于所述本发明的稠环芳香族化合物。特别是,所述稠环芳香族化合物此外还可优选包含其它化合物。从液相处理根据本发明的稠环芳香族化合物,例如通过旋涂或通过印刷方法进行处理,需要根据本发明的化合物的制剂。这些制剂可以例如是溶液、分散体或乳液。出于这个目的,可优选使用两种或更多种溶剂的混合物。合适并且优选的溶剂例如是甲苯,苯甲醚,邻二甲苯、间二甲苯或对二甲苯,苯甲酸甲酯,均三甲苯,萘满,邻二甲氧基苯,四氢呋喃,甲基四氢呋喃,四氢吡喃,氯苯,二噁烷,苯氧基甲苯,特别是3-苯氧基甲苯,(-)-葑酮,1,2,3,5-四甲基苯,1,2,4,5-四甲基苯,1-甲基萘,2-甲基苯并噻唑,2-苯氧基乙醇,2-吡咯烷酮,3-甲基苯甲醚,4-甲基苯甲醚,3,4-二甲基苯甲醚,3,5-二甲基苯甲醚,苯乙酮,α-萜品醇,苯并噻唑,苯甲酸丁酯,异丙苯,环己醇,环己酮,环己基苯,十氢化萘,十二烷基苯,苯甲酸乙酯,茚满,苯甲酸甲酯,1-甲基吡咯烷酮,对甲基异丙基苯,苯乙醚,1,4-二异丙基苯,二苄醚,二乙二醇丁基甲基醚,三乙二醇丁基甲基醚,二乙二醇二丁基醚,三乙二醇二甲基醚,二乙二醇单丁基醚,三丙二醇二甲基醚,四乙二醇二甲基醚,2-异丙基萘,戊苯,己苯,庚苯,辛苯,1,1-双(3,4-二甲基苯基)乙烷,或这些溶剂的混合物。
[0053]
进一步地,所述的有机层还包括电子注入层、电子输送层、空穴阻挡层、电子阻挡层、空穴输送层、空穴注入层、发光层中的一种。
[0054]
进一步地,所述的发光层包括掺杂物和发光主体,所述的掺杂物包括所述的苯并菲衍生物,所述的发光主体为所述的苯并菲衍生物、萘、蒽、芘、苝、菲、荧蒽、苯并蒽和并五苯及其衍生物中的一种。
[0055]
进一步地,所述的掺杂物与发光主体的质量比为1:99~50:50。
[0056]
与现有技术相比,本发明的有益效果为:
[0057]
本发明所述的苯并菲衍生物具有吲哚、苯并呋喃、苯并噻吩并苯并菲的稠环结构的新型有机电致发光化合物,是在苯并菲的基础上增大分子内的电子密度,同时调整取代基团增大分子位阻、减弱分子间的π-π相互作用,提高分子的内量子效率,并且具有与现有
的化合物相比更短的发光波长。同时,所述的苯并菲衍生物阻碍有机分子间激-激复合物的生成、增加内部电子密度和稳定性,因此,利用本发明所述的苯并菲衍生物制备有机电致发光器件的效果和寿命明显提高,此外,所述的苯并菲衍生物改善了对溶液的溶解度而解决以往蓝光材料所具有的工序的生产性以及费用的问题,并且在原有的工序中不是其蒸镀工序,而是在其他溶液工序中也可以用于制备发光层。
附图说明
[0058]
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0059]
图1是本发明的有机电致发光装置的一个底部发光例子的示意图;
[0060]
图2是本发明的有机电致发光装置的一个顶部发光例子的示意图。
[0061]
附图标记
[0062]
1-基板、2-阳极、3-孔穴注入层、4-孔穴传输/电子阻挡层、5-发光层、6-孔穴阻挡/电子传输层、7-电子注入层、8-阴极。
具体实施方式
[0063]
为使本发明的目的、技术方案和优点更加清楚,下面将对本发明的技术方案进行详细的描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所得到的所有其它实施方式,都属于本发明所保护的范围。
[0064]
下述对oled材料及元件进行性能测试的测试仪器及方法如下:
[0065]
oled元件性能检测条件:
[0066]
亮度和色度坐标:使用光谱扫描仪photoresearch pr-715测试;
[0067]
电流密度和起亮电压:使用数字源表keithley 2420测试;
[0068]
功率效率:使用newport 1931-c测试;
[0069]
寿命测试:使用lts-1004ac寿命测试装置。
[0070]
实施例1
[0071]
化合物cjhb271的制备方法,包括如下步骤:
[0072]
第一步:化合物int.-1的制备
[0073][0074]
在氮气保护下,24.5g(127.0mmol)的对氯溴苯溶于300ml干燥的四氢呋喃中,用液氮降温至-78℃,缓慢滴加入56.0ml的2.5m正丁基锂正己烷溶液,搅拌反应1小时,滴加入19.5g(140.0mmol)的对氯苯腈溶于50ml干燥四氢呋喃的溶液,升至室温搅拌反应2小时,加
入50ml的饱和氯化铵水溶液,搅拌反应30分钟,分出有机相,水相用乙酸乙酯萃取,收集有机相干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到27.7g白色固体,收率87%。
[0075]
第二步:化合物int.-2的制备
[0076][0077]
在氮气保护下,12.6g(50.0mmol)第一步制备的int.-1溶解于200ml的二氯甲烷中,冰水浴降温到0℃,加入20.0g的四溴化碳和12.5g的亚磷酸三异丙酯,搅拌反应30分钟,升到室温反应1小时,滴加入50ml的1n稀盐酸水溶液,搅拌30分钟,分出有机相,水相用二氯甲烷萃取,收集有机相,干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到白色固体int.-2,收率92%。
[0078]
第三步:化合物int.-3的制备
[0079][0080]
取20.0g(50.0mmol)第二步制备的中间体int.-2与100ml的1,4-二氧六环混合,在氮气保护下,加入110.0mmol的苯并呋喃-2-硼酸或苯并噻吩-2-硼酸或1-取代吲哚-2-硼酸或1,1-取代的茚-2-硼酸和112.5mg的醋酸钯催化剂、410.5mg的2-双环己基膦-2
’
,6
’-
二甲氧基联苯以及80.0mmol的水合磷酸钾,再加入40ml的水,升温至60℃搅拌反应12小时,冷却到室温,加入100ml的乙酸乙酯和100ml的水,分出有机相,水相再用乙酸乙酯萃取,收集有机相再用饱和盐水洗,有机相干燥过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到白色固体int.-3,收率60~65%。
[0081]
第四步:化合物int.-4的制备
[0082][0083]
20.0mmol第三步制备的中间体int-3和100ml的氯仿、20ml的硝基甲烷混合,再加入1.6g的无水氯化铁,加热回流反应24小时,冷却到室温,加入50ml稀盐酸水溶液,用二氯甲烷萃取,收集有机相,干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到中间体int.-4,收率55~65%。
[0084]
第五步:化合物cjhb271的制备
[0085][0086]
将5.0mmol第四步制备的中间体int.-4分散在60ml的甲苯中,加入12.0mmol的二苯胺,再加入1.5g(15.0mmol)的叔丁醇钠、55.0mg(0.05mmol)的pd2(dba)3chcl3催化剂和0.1ml的10%叔丁基磷甲苯溶液,升温至100℃搅拌反应12小时,反应结束后,加入50ml的水,分出有机相,水相用甲苯-thf萃取,收集有机相干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,再用二氯甲烷和乙醇重结晶,得到cjhb271,黄色固体。实验数据如表1所示:
[0087]
表1
[0088][0089]
实施例2
[0090]
化合物cjhb387的制备方法,包括如下步骤:
[0091]
第一步:化合物int.-5的制备
[0092][0093]
在氮气保护下,12.8g(50.0mmol)的2,7-二甲氧基氧杂蒽-9-酮溶解于200ml的二氯甲烷中,冰水浴降温到0℃,加入20.0g的四溴化碳和12.5g的亚磷酸三异丙酯,搅拌反应30分钟,升到室温反应1小时,滴加入50ml的1n稀盐酸水溶液,搅拌30分钟,分出有机相,水相用二氯甲烷萃取,收集有机相,干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到白色固体int.-5,收率94%。
[0094]
第二步:化合物int.-6的制备
[0095][0096]
参照实施例1第三步的制备方法,仅将实施例1第三步中的int.-2替换为中间体int.-5,制备中间体int.-6,收率80~90%。
[0097]
第三步:化合物int.-7的制备
[0098][0099]
参照实施例1第四步的制备方法,仅将实施例1第四步中的int.-3替换为中间体int.-6,于室温搅拌反应12小时,制备中间体int.-7,收率65~75%。
[0100]
第四步:化合物int.-8的制备
[0101][0102]
在氮气保护下,20.0mmol的中间体int.-7溶解于120ml干燥的二氯甲烷中,用液氮降温至-40℃,滴加入40.0mmol的三溴化硼溶于二氯甲烷的溶液,搅拌反应2小时,升到室温,滴加入50ml的1n稀盐酸水溶液,搅拌1小时,分出有机相,水相用二氯甲烷萃取,有机相干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到固体int.-8,收率90~94%。
[0103]
第五步:化合物int.-9的制备
[0104][0105]
在氮气保护下,20.0mmol的中间体int.-8溶解于120ml干燥的二氯甲烷中,加入60.0mmol的吡啶,用冰水浴降温至0℃,滴加入48.0mmol的三氟甲基磺酸酐溶于二氯甲烷的溶液,搅拌反应2小时,升到室温搅拌反应2小时,加入50ml的1n稀盐酸水溶液,分出有机相,水相用二氯甲烷萃取,有机相干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到固体int.-9,收率92~96%。
[0106]
第六步:化合物cjhb387的制备
[0107][0108]
将5.0mmol第五步制备的中间体int.-9分散在60ml的甲苯中,加入12.0mmol的4,4
’-
二甲基二苯胺,再加入2.0g(20.0mmol)的叔丁醇钠、55.0mg(0.05mmol)的pd2(dba)3chcl3催化剂和0.1ml的10%叔丁基磷甲苯溶液,升温至100℃搅拌反应12小时,反应结束后,加入50ml的水,分出有机相,水相用乙酸乙酯-thf萃取,收集有机相干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,再用thf-甲苯重结晶,得到cjhb387,黄色固体。
[0109]
实验数据如表2所示。
[0110]
表2
[0111][0112]
实施例3
[0113]
化合物cjhb270~cjhb324、cjhb386、cjhb388~cjhb403的制备:
[0114]
参照实施例1~2的制备方法,仅将不同的肿胺替换实施例1中第五步的二苯胺或实施例2中第六步的4,4
’-
二甲基二苯胺,其他实验参数进行常规调整。
[0115]
实施例4
[0116]
化合物cjhb336的制备:
[0117]
第一步:化合物int.-11的制备
[0118][0119]
6.6mmol的中间体int.-10(参照实施例1的合成方法,仅将第一步中的对氯溴苯替
换为溴苯制备中间体int.-10)分散于100ml干燥的thf中,在氮气保护下,用液氮降温至-78℃,滴加入3.2ml的2.5m叔丁基锂,搅拌反应1小时,滴加入10.0mmol的硼酸三甲酯,反应1小时后,加入50ml的2n稀盐酸水溶液,分出有机相,水相用乙酸乙酯萃取,收集有机相干燥,过滤,滤液减压浓缩干,用石油醚洗涤,过滤,得到白色固体,收率65~75%。
[0120]
第二步:化合物cjhb336的制备
[0121][0122]
取5.0mmol第一步制备的中间体int.-10与40ml的甲苯混合,在氮气保护下,加入4.0mmol的2-氯-4,6-二苯基-1,3,5-三嗪、10.0mmol的无水碳酸钾和58.0mg的pd(pph3)4催化剂,再加入20ml的乙醇和20ml的水,升温至回流搅拌反应12小时,冷却到室温,加入50ml的二氯甲烷和100ml的水,分出有机相,水相再用二氯甲烷萃取,收集有机相再用饱和盐水洗,有机相干燥过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到白色固体cjhb336,收率75~86%。
[0123]
实验数据如表3所示。
[0124]
表3
[0125][0126]
实施例5
[0127]
化合物cjhb325~cjhb335、cjhb337~cjhb385的制备:
[0128]
参照实施例4的制备方法,仅将不同的卤代物替换实施例4中第二步的2-氯-4,6-二苯基-1,3,5-三嗪,其他实验参数进行常规调整。
[0129]
实施例6
[0130]
化合物cjhb413的制备方法,包括如下步骤:
[0131]
第一步:化合物int.-13的制备
[0132][0133]
取22.0g(50.0mmol)的中间体int.-12(参照实施例2的合成方法制备)与100ml的1,4-二氧六环混合,在氮气保护下,加入110.0mmol的苯并呋喃-3-硼酸或苯并噻吩-3-硼酸或1-取代吲哚-3-硼酸或1,1-取代的茚-3-硼酸和112.5mg的醋酸钯催化剂、410.5mg的2-双
环己基膦-2
’
,6
’-
二甲氧基联苯以及80.0mmol的水合磷酸钾,再加入40ml的水,升温至60℃搅拌反应12小时,冷却到室温,加入100ml的乙酸乙酯和50ml的水,分出有机相,水相再用乙酸乙酯萃取,收集有机相再用饱和盐水洗,有机相干燥过滤,滤液减压浓缩干,用硅胶柱分离纯化,得到中间体int.-13,收率70~80%。
[0134]
第二步:化合物int.-14的制备
[0135][0136]
在氮气保护下,10.5mmol第一步制备的中间体int.-13溶解于200ml的二氯甲烷中,加入63.0mmol的碘,再加入10ml的环氧丙烷,用254nm的紫外灯照射,搅拌反应12小时,减压浓缩干,加入100ml的甲醇分散,过滤,滤饼用硅胶柱分离纯化,得到黄色固体中间体int.-14,收率85~95%。
[0137]
第三步:化合物int.-15的制备
[0138][0139]
参照实施例2第四步的合成方法,仅将实施例2第四步中的中间体int.-7替换为中间体int.-13,制备得到中间体int.-15,收率92~96%。
[0140]
第四步:化合物int.-16的制备
[0141][0142]
参照实施例2第五步的合成方法,仅将实施例2第五步中的中间体int.-8替换为中间体int.-15,制备得到中间体int.-16,收率85~90%。
[0143]
第五步:化合物cjhb413的制备
[0144][0145]
将5.0mmol第四步制备的中间体int.-16分散在60ml的甲苯中,加入12.0mmol的4,4
’-
二甲基二苯胺,再加入2.0g(20.0mmol)的叔丁醇钠、55.0mg(0.05mmol)的pd2(dba)3chcl3催化剂和0.1mmol的xanphos,升温至90℃搅拌反应12小时,反应结束后,加入50ml的水,分出有机相,水相用乙酸乙酯-thf萃取,收集有机相干燥,过滤,滤液减压浓缩干,用硅胶柱分离纯化,再用thf-甲苯重结晶,得到cjhb413,黄色固体。
[0146]
实验数据如表4所示:
[0147]
表4
[0148][0149]
实施例7
[0150]
化合物cjhb404~cjhb412、cjhb414~cjhb429的制备:
[0151]
参照实施例6的制备方法,仅将不同的肿胺替换实施例6中第五步的4,4
’-
二甲基二苯胺,其他实验参数进行常规调整。
[0152]
实施例8
[0153]
化合物cjhb430~cjhb480的制备:
[0154]
参照实施例4的制备方法,仅将不同的卤代物替换实施例4中第二步的2-氯-4,6-二苯基-1,3,5-三嗪,其他实验参数进行常规调整。
[0155]
试验例1
[0156]
如图1所示,一种oled元件,所示的oled元件的制备方法包括如下步骤:
[0157]
1)将涂布了ito导电层的玻璃基片在清洗剂中超声处理30分钟,在去离子水中冲洗,在丙酮/乙醇混合溶剂中超声30分钟,在洁净的环境下烘烤至完全干燥,用紫外光清洗机照射10分钟,并用低能阳离子束轰击表面。
[0158]
2)把上述处理好的ito玻璃基片置于真空腔内,抽真空至1
×
10-5
~9
×
10-3
pa,在上述阳极层膜上继续分别蒸镀化合物dntpd作为空穴注入层,蒸镀膜厚为在上述空穴注入层膜上继续蒸镀npd为空穴传输层,蒸镀膜厚为
[0159]
3)在空穴传输层上继续蒸镀一层化合物ht202作为电子阻挡层,蒸镀膜厚为
[0160]
4)在电子阻挡层上继续蒸镀一层实施例1-8制备的苯并菲衍生物cjhb271~cjhb480中的一种和bh011作为有机发光层,其中,bh011为主体材料和本发明所述的苯并菲衍生物为掺杂材料,所述的的苯并菲衍生物在bh011中的掺杂浓度为1~20%,蒸镀膜厚为
[0161]
5)在上述有机发光层上再继续蒸镀一层化合物tpbi作为空穴阻挡层,蒸镀膜厚为
[0162]
6)在上述空穴阻挡层上再继续蒸镀一层化合物liq和et205作为器件的电子传输层,其中,liq和et205的质量比为1:1,蒸镀膜厚为
[0163]
7)在上述空穴阻挡层上再继续蒸镀一层化合物lif作为器件的电子传输层,蒸镀膜厚为最后,在上述电子传输层之上蒸镀金属铝作为元件的阴极层,蒸镀膜厚为
[0164]
其中,试验例1中所使用的化合物结构式如下所示:
[0165][0166]
试验样品1
[0167]
试验例1中的苯并菲衍生物为cjhb271-1,其他按照试验例1的方法制备的发光元件。
[0168]
试验样品2
[0169]
试验例1中的苯并菲衍生物为cjhb271-4,其他按照试验例1的方法制备的发光元件。
[0170]
试验样品3
[0171]
试验例1中的苯并菲衍生物为cjhb387-1,其他按照试验例1的方法制备的发光元件。
[0172]
试验样品4
[0173]
试验例1中的苯并菲衍生物为cjhb387-4,其他按照试验例1的方法制备的发光元件。
[0174]
试验样品5
[0175]
试验例1中的苯并菲衍生物为cjhb336-2,其他按照试验例1的方法制备的发光元件。
[0176]
试验样品6
[0177]
试验例1中的苯并菲衍生物为cjhb413-2,其他按照试验例1的方法制备的发光元件。
[0178]
试验样品7
[0179]
试验例1中的苯并菲衍生物为cjhb413-5,其他按照试验例1的方法制备的发光元
件。
[0180]
对照样品1
[0181]
试验例1中的苯并菲衍生物替换为化合物bd10,其他按照试验例1的方法制备的发光元件,其中,化合物bd10的结构式如下所示:
[0182][0183]
将试验样品1-7和对照样品1的发光元件的性能进行检测,结果如表5所示。
[0184]
表5
[0185][0186]
上表5中可以看出,在电流密度为10ma/cm2条件下的驱动电压、半峰宽fwhm、电流效率以及在亮度为1000cd/m2初始条件下的器件寿命lt90%的数据是针对对照样品1归一化。
[0187]
结论:从性能测试结果表5可以看到,本发明的化合物作为蓝光染料获得了深蓝光有机电致发光器件,相较于采用bd10作为蓝光掺杂材料的有机电致发光器件,元件的电流效率更高、驱动电压降低,而且器件初始亮度在1000cd/m2的条件下,器件的lt90%寿命也有了很大的改善。
[0188]
图1和2是本发明的有机电致发光装置的一个底部发光例子的示意图与本发明的有机电致发光装置的一个顶部发光例子的示意图。
[0189]
产业上的应用可能性
[0190]
本发明的有机电致发光装置可以在壁挂电视、平板显示器、照明等的平面发光体、
复印机、打印机、液晶显示器的背光源或计量仪器类等的光源,显示板、标识灯等中应用。
[0191]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本发明的保护范围之内。因此,本发明的保护范围应以所述权利要求的保护范围为准。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips