
 商标分类
商标分类  商标转让
商标转让 
一种人iPS细胞基因编辑和筛选方法与流程
 2021-02-02 06:02:06|
2021-02-02 06:02:06| 381|
381| 起点商标网
起点商标网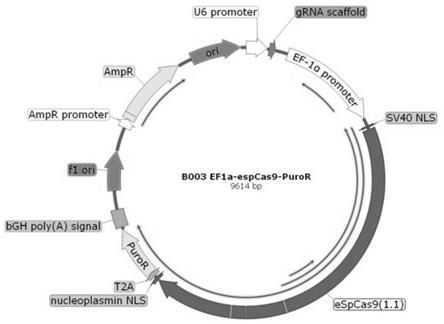
一种人ips细胞基因编辑和筛选方法
技术领域
[0001]
本发明涉及对细胞进行基因编辑和筛选的方法,尤其是一种人ips细胞基因编辑和筛选方法,具体地说,进行crispr/cas9介导的基因敲除的方法和利用抗性筛选进行富集,再进行细胞筛选的方法。
背景技术:
[0002]
crispr-cas9是细菌和古细菌在长期演化过程中形成的一种适应性免疫防御,可用来对抗入侵的病毒及外源dna。而crispr-cas9基因编辑技术,则是对靶向基因进行特定dna修饰的技术,这项技术也是用于基因编辑中前沿的方法。cas9酶蛋白可在grna的引导下在基因组特定位置产生双链断裂,从而激发细胞内的dna损伤修复机制,胞内修复 dna双链断裂的机制主要有两条:一条是非同源末端连接途径(non-homologous endjoining,nhej),在该途径中细胞对dna断裂处进行部分酶切,使之产生粘性末端,再将两个末端连接起来,这个过程中会导致随机数目碱基丢失,使得该相应基因缺乏几个氨基酸或者发生移码突变甚至大片段的基因丢失而被敲除。另一条是同源重组途径 (homology-directed recombination,hdr),若dna损伤时的同时恰好遇到与该损伤位点同源性很高的另一条完整的dna时,损伤位点会以同源的dna为模板进行损伤修复,若同源基因中被人为引入了其它基因、元件或者点突变则可凭此修改细胞基因组。
[0003]
诱导性多能干(ips)细胞是指通过导入特定的转录因子将终末分化的体细胞恢复到全能性状态所形成的干细胞系。ips细胞具有无限增殖潜能和三胚层分化能力,可以分化为各种类型的成熟细胞。利用crispr/cas9技术对ips细胞进行基因编辑可以通过单克隆挑选和扩增,建立稳定的细胞系,长久的保留基因改变,解决成熟细胞基因编辑后增殖能力有限,无法稳定保存,无法进行单克隆纯化的问题,并且其来源的分化细胞也携带有基因改变,是一种简便快捷制备基因修饰细胞的方案。
[0004]
但人类的ips细胞属于难编辑细胞,相对于人成熟细胞,人ips细胞更难导入并表达外源基因,转化效率、基因编辑效率都较低,编辑效率一般在1%以下,进行单克隆筛选工作量大,难以进行。为了筛选出经过基因编辑的细胞主要使用流式分选技术,或稳转抗性筛选技术,来富集出经过基因编辑的ips细胞,再进行单克隆挑取和建系扩增。流式分选需要昂贵的流式分选仪器和极高的实验条件,且所编辑的基因需在ips细胞上为阳性表达以便于筛选出编辑后表达阴性的细胞,对于在ips细胞中不表达或低表达的基因则无因无法进行区分而无法筛选,为此需要在编辑的同时插入组成性表达的抗性或荧光蛋白基因,以此作为筛选标记,这需要在编辑位置插入较长的外源基因序列,对人类基因组可能带来未知的影响。
技术实现要素:
[0005]
本发明所要解决的技术问题是,提供一种不需要使用流式分选,不在基因组内插入外源筛选标志基因的人ips细胞基因编辑和筛选方法。
[0006]
为了解决上述技术问题,本发明采用的技术方案是:一种人ips细胞基因编辑和筛选方法,包括以下步骤:(1)将编辑质粒通过电转方法导入ips细胞;(2)通过抗性筛选富集产生基因编辑的细胞;(3)低密度传代,挑取单克隆细胞集落进行扩大培养和鉴定。
[0007]
所述电转方法为:将ips细胞使用accutase分散为单细胞,重悬于20ul lonza p3 电转buffer中,加入基因编辑质粒,在lonza 4d-nucleofector上使用ca-137程序进行电转。每次电转的ips细胞数量为1*105到5*105。
[0008]
所述编辑质粒为ef1α-espcas9-puror(seq id no:1),质粒上含ef1a启动子(seqid no:2),其能在ips细胞内启动通过espcas9(seq id no:3)和puror(seq id no: 4)两种蛋白编码序列的转录并在ips细胞内表达这两种蛋白。
[0009]
所述编辑质粒的添加剂量为100-1000ng。
[0010]
所述步骤(2)包括:1)将电转后细胞悬于含rock抑制剂的培养基中接种到feeder 或基质上,二氧化碳细胞培养箱中进行细胞恢复培养;2)加入筛选物质进行抗性筛选;
[0011]
3)抗性筛选结束后更换为含rock抑制剂培养基进行细胞培养扩增。
[0012]
所述细胞恢复培养时间为2-24小时,抗性筛选时间为12-24小时。所述筛选物质为嘌呤霉素,添加终浓度为0.4-2ug/ml。所述抗性筛选结束后,培养基中rock抑制剂的添加时长为0-5天。
[0013]
优选,每次电转的ips细胞数量为2*105,编辑质粒的添加剂量为300ng,细胞恢复培养时间为6小时,抗性筛选时间为18小时,筛选物质嘌呤霉素添加终浓度为0.8ug/ml,养基中rock抑制剂的添加时长为2天。
[0014]
本发明的有益效果是:无需使用流式分选仪器,即可实现人ips细胞基因敲除编辑。可以对ips细胞不表达的基因进行基因编辑敲除。无需在基因组上插入组成性表达的筛选抗性,避免大片段外源基因插入对基因组稳定性的影响。基因编辑率大幅提高,便于单克隆挑选和建系。
附图说明
[0015]
图1是实施例1中构建的用于人ips细胞基因编辑的质粒, hu6-sgrna-ef1a-espcas9(1.1)-2a-puro。
[0016]
图2是实施例3中基因编辑后b2m完全敲除的ips细胞系ek33的b2m基因测序结果。
[0017]
图3是实施例3中基因编辑后b2m完全敲除的ips细胞系ek33的流式检测结果。
具体实施方式
[0018]
以下结合具体实施例对上述方案做进一步说明。应理解,这些实施例是用于说明本发明而不限于本发明的范围。实施例中采用的实施条件可以根据具体厂家的条件做进行调整,未注明的实施条件通常为常规实验中的条件。
[0019]
本发明的人ips细胞基因编辑和筛选方法,包括以下步骤:(1)将编辑质粒通过电转方法导入ips细胞;(2)通过抗性筛选富集产生基因编辑的细胞;(3)低密度传代,挑取单克隆细胞集落进行扩大培养和鉴定。
[0020]
所述电转方法为:将ips细胞使用accutase分散为单细胞,重悬于20ul lonza p3 电转buffer中,加入基因编辑质粒,在lonza 4d-nucleofector上使用ca-137程序进行电
转。每次电转的ips细胞数量为1*105到5*105。
[0021]
所述编辑质粒为ef1α-espcas9-puror(seq id no:1),质粒上含ef1a启动子(seqid no:2),其能在ips细胞内启动通过espcas9(seq id no:3)和puror(seq id no: 4)两种蛋白编码序列的转录并在ips细胞内表达这两种蛋白。所述编辑质粒的添加剂量为100-1000ng。
[0022]
所述步骤(2)包括:1)将电转后细胞悬于含rock抑制剂的培养基中接种到feeder 或基质上,二氧化碳细胞培养箱中进行细胞恢复培养;2)加入筛选物质进行抗性筛选; 3)抗性筛选结束后更换为含rock抑制剂培养基进行细胞培养扩增。
[0023]
所述细胞恢复培养时间为2-24小时,抗性筛选时间为12-24小时。所述筛选物质为嘌呤霉素,添加终浓度为0.4-2ug/ml。所述抗性筛选结束后,培养基中rock抑制剂的添加时长为0-5天。
[0024]
优选,每次电转的ips细胞数量为2*105,编辑质粒的添加剂量为300ng,细胞恢复培养时间为6小时,抗性筛选时间为18小时,筛选物质嘌呤霉素添加终浓度为0.8ug/ml,养基中rock抑制剂的添加时长为2天。
[0025]
本发明由ef1α启动子启动能在人ips细胞内同时启动cas9和筛选抗性蛋白表达的游离型质粒,此质粒进入人ips细胞中后,可在细胞中短暂而稳定的同时表达cas9蛋白和抗性蛋白,在质粒转录表达最旺盛的时间段,在培养环境中添加筛选物,获得外源质粒并表达抗性蛋白的ips细胞得以存活,同时表达的cas9蛋白在sgrna的引导下在基因组目的序列位置进行双链切割进行基因编辑,而未获得外源质粒或质粒未表达的细胞则因不含有抗性而死亡。经过抗性筛选可大幅提高基因编辑细胞的比例,从文献报道的不足1%,提升到40%以上,之后再进行低密度传代和单克隆细胞集落挑选,即可获得基因编辑后的细胞系。利用通过对瞬时表达质粒上抗性基因进行筛选,提高存活细胞中编辑效率,在通过单克隆筛选挑选基因编辑后细胞系的基因编辑和筛选方案,避免流式细胞分选仪的使用或外源基因插入到细胞基因组中。
[0026]
实施例1用于人ips细胞基因编辑质粒的构建
[0027]
构建一个由ef1a启动子同时引导高特异性cas9蛋白基因和嘌呤霉素抗性基因表达,并具有hu6启动子启动sgrna合成的一体化质粒 (hu6-sgrna-ef1a-espcas9(1.1)-2a-puro),质粒主体框架为#64139质粒,espcas9(1.1) 来源于#71814质粒,ef1a启动子序列来源于#60599质粒。(所用质粒采购自addgene,内切酶采购自thermo,一步法克隆试剂盒采购子诺唯赞)
[0028]
1.1更改为espcas9(1.1),构建质粒b002
[0029]
1.1.1#64139质粒线性化
[0030]
64139质粒1ugbuffer r2ulbsm i1ulecor v1ulddh2oupto 20ul
[0031]
37℃酶切过夜,80℃灭活20分钟
[0032]
1.1.2插入片断pcr
[0033]
#71814质粒1ugecor i buffer2ulecor i1ulddh2oupto 20ul
[0034]
37℃酶切过夜,80℃灭活20分钟,取1ul酶切产物加入到1mlddh2o中稀释模板,使用下列引物进行pcr扩增,产物长度1836bp
[0035]
hscas9-ftgaggaaaacgaggacattctggaagatseq id no:5hscas9-rgcagttcgccggcagaggseq id no:6
[0036]
1.1.3一步法克隆
[0037]
64139酶切产物2ulpcr产物2ul5
×
ce ii buffer4ulexnase2ulddh2o10ul
[0038]
37℃孵育30分钟,立刻进行转化
[0039]
1.1.4转化
[0040]
产物转化进dh5a感受态细胞,在氨苄青霉素lb平板上涂板,37℃过夜培养,挑取单菌落小摇菌液进行测序验证,测序正确的菌液建库冻存、扩大培养提取质粒。
[0041]
1.2更换ef1a启动子(b003)
[0042]
1.2.1质粒b002线性化
[0043]
b002质粒1ugbuffer ango2ulncoi1ulxbai1ulddh2oupto 20ul
[0044]
37℃酶切过夜,65℃灭活20分钟
[0045]
1.2.2插入片断pcr
[0046]
#60599质粒1ugecor i buffer2ulecor i1ulddh2oupto 20ul
[0047]
37℃酶切过夜,65℃灭活20分钟,取1ul酶切产物加入到1mlddh2o中稀释模板,使用下列引物进行pcr扩增,产物长度1295bp
[0048]
ef1a-ftgcagacaaatggctctagagagaggaatctttgcagctaatggaccseq id no:7ef1a-rcgtggtccttatagtccatggtggcgccgccaccgctaattctcacseq id no:8
[0049]
1.2.3一步法克隆
[0050]
b002酶切产物2ul
pcr产物2ul5
×
ce ii buffer4ulexnase2ulddh2o10ul
[0051]
37℃孵育30分钟,立刻进行转化
[0052]
1.2.4转化
[0053]
产物转化进dh5a感受态细胞,在氨苄青霉素lb平板上涂板,37℃过夜培养,挑取单菌落小摇菌液进行测序验证,测序正确的菌液建库冻存、扩大培养提取质粒。
[0054]
构建的质粒图谱如图1所示。
[0055]
实施例2人ips细胞b2m基因敲除
[0056]
内切酶、连接酶购自thermo,mtesr1、cloner、accutase、metrigel购自stemcell,电转试剂购自lonza。
[0057]
2.1插入b2m sgrna序列,构造b2m敲除工作质粒(c003),
[0058]
2.1.1 sgrna序列引物合成:合成下列引物
[0059]
b2msg-fcaccgcgcgagcacagctaaggccaseq id no:9b2msg-raaactggccttagctgtgctcgcgcseq id no:10
[0060]
ddh2o溶解引物到10um,
[0061]
2.1.2 b003质粒线性化
[0062]
b003质粒1ugfastdigest bpii1ul10
×
fastdigest buffer2ulddh2oupto 20ul
[0063]
37℃处理30分钟
[0064]
2.1.3 sgrna序列退火稀释
[0065]
b2msg-f(10um)4.5ulb2msg-r(10um)4.5ul10
×
pcr buffer1ul
[0066]
将装有混合物的pcr管放入装有沸水的烧杯中,室温放置到沸水自然冷却。
[0067]
88ul ddh2o中加入2ul退火后的sgrna混匀,稀释到100nm。
[0068]
2.1.4质粒连接
[0069]
线性化b003(50ng/ul)2ulsgrna退火引物稀释液(100nm)2ul10
×
t4 buffer2ulfast t4 ligase1ulddh2o13ul
[0070]
混匀后室温放置10分钟
[0071]
2.1.5转化
[0072]
连接产物转化dh5a感受态细胞,在氨苄青霉素lb平板上涂板,37℃过夜培养,挑取
单菌落小摇菌液,进行测序验证,测序正确的菌液建库冻存、扩大培养提取质粒
[0073]
2.2电转质粒
[0074]
2.2.1 ips单细胞悬液制备
[0075]
人ips细胞在6孔板内生长到覆盖度约70%时,吸弃培养基,每孔用1ml dpbs冲洗残留培养基后吸弃,加入1ml accutase 37℃培养6分钟,加入2ml dpbs终止反应,反复吹打细胞到形成单细胞悬液,进行细胞计数。
[0076]
2.2.2质粒电转
[0077]
取2
×
105个ips细胞,200g离心去除上清,加入20ul p3电转液(lonza)重悬,加入300ng c003质粒,使用lonza 4d-nucleofector进行电转,电转程序ca-137。电转结束后立即向电转杯中加入100ul mtesr1(含10%cloner)吹打均匀,37℃培养5分钟后转到含2ml mtesr1(含10%cloner)的metrigel包被的6孔培养板中37℃培养。
[0078]
2.3抗性筛选
[0079]
接种6小时后,加入16ul 100ug/ml的嘌呤霉素,使终浓度达到0.8ug/ml,摇匀后继续37℃培养,18小时后,吸弃培养基,加入2ml mtesr1(含10%cloner)培养基,37 ℃培养2天,之后每日更换新鲜mtesr1培养基,适当时进行传代培养,并建库冻存。
[0080]
2.4单克隆筛选
[0081]
2.4.1低密度传代
[0082]
当ips细胞生长到覆盖70%孔底时,使用accutase收集单细胞,按1:100,1:200 和1:500比例分别接种到含mtesr1(含10%cloner)metrigel包被的6孔培养板中, 37℃培养,接种2天后更换为mtesr1培养基37℃继续培养,每日更换培养基直到单克隆细胞集落形成。
[0083]
2.4.2单克隆挑取
[0084]
24孔板用matrigel包被,每孔加入600ul mtesr 1培养基,倒置相差显微镜4倍镜下,挑取单克隆细胞集落到培养孔中,用1ml移液器吹洗5-6次吹散细胞,摇匀后37℃培养,第二天更换为mtesr 1培养基,之后每日更换培养基到细胞覆盖度约70%后传代到 6孔培养板中,并进行后续传代培养和冻存。
[0085]
实施例3编辑结果鉴定
[0086]
3.1细胞系dna提取
[0087]
当ips细胞生长到覆盖70%孔底时,吸弃培养基,每孔加入1ml dpbs冲洗残留培养基后吸弃,每孔加入1ml 0.5mm edta溶液,37℃培养10分钟,用1ml移液器反复吹打冲洗板底,转移全部液体到1.5ml离心管中,300g离心5分钟,去除上清。使用tiangen 全血基因组dna提取试剂盒,依说明操作提取dna。
[0088]
3.2编辑结果测序检测
[0089]
使用引物b2m-f:cagcaaggacatagggaggaac(seq id no:11)和b2m-r: caccaaggagaacttggagaag(seq id no:12)进行扩增,产物进行sanger法测序。其中编号ek33的细胞系测序结果如图2所示,结果显示基因组在sgrna识别位置被cas9蛋白切断后丢失2个碱基并随机插入了6个碱基,在读码框内插入了终止密码子,提前终止了蛋白表达。
[0090]
挑取单克隆细胞集落40个,共35个建系并测序成功,其中15个细胞系发生明显的基因序列改变或移码引起乱峰,未发生基因编辑的细胞系20个,基因编辑发生率42.8%。
[0091]
3.3流式检测
[0092]
用accutas处理回收细胞系为单细胞悬液,200g离心去除上清后重悬于dpbs,使用 fitc mouse anti-humanβ2-microglobulin抗体(bd 551338)fitc mouse igm,κisotype control抗体(bd 555583)分别进行标记,使用流式细胞仪收集数据后进行分析,检测细胞便面b2m分布。未经基因编辑的ips细胞系和ek33细胞系流式分析结果如图3所示,未编辑ips细胞系与同型对照相比,b2m分布呈阳性,ek16、ek26、ek33三个细胞系表面低表达b2m蛋白。
[0093]
3.4基因编辑脱靶检测
[0094][0095][0096]
利用表中引物,分别对8个潜在脱靶位点进行pcr扩增,之后进行sanger法测序检测,ips细胞系在基因编辑前后,上述8个位置基因序列未发生改变,与数据库序列一致,基因编辑过程中未发生脱靶。
[0097]
对比例1电转质粒48-72小时进行抗性筛选取2
×
105个ips细胞,200g离心去除上清,加入20ul p3电转液(lonza)重悬,加入300ng c003质粒,使用lonza 4d-nucleofector进行电转,电转程序ca-137。电转结束后立即向电转杯中加入100ul mtesr1(含10%cloner)吹打均匀,37℃培养5分钟后转到含2ml mtesr1(含10%cloner)的metrigel包被的6孔培养板中37℃培养。接种48 小时后,吸弃孔中培养基,更换为2ml新鲜mtesr 1培养基,加入16ul 100ug/ml的嘌呤霉素,使终浓度达到0.8ug/ml,摇匀后继续37℃培养,24小时后,吸弃培养基,加入 2ml mtesr1(含10%cloner)培养基,37℃培养2天。全部细胞悬浮死亡,
无细胞存活。
[0098]
对比例2不进行抗性筛选
[0099]
取2
×
105个ips细胞,200g离心去除上清,加入20ul p3电转液(lonza)重悬,加入300ng c003质粒,使用lonza 4d-nucleofector进行电转,电转程序ca-137。电转结束后立即向电转杯中加入100ul mtesr1(含10%cloner)吹打均匀,37℃培养5分钟后转到含2ml mtesr1(含10%cloner)的metrigel包被的6孔培养板中37℃培养。之后每日更换新鲜mtesr1培养基,适当时进行传代培养,并建库冻存。
[0100]
进行低密度传代和单克隆挑取,分别扩大培养,共挑取单克隆细胞系33个,全部细胞系提取dna进行测序,未发现基因序列发生改变的细胞系。
[0101]
对比例3使用#64139质粒进行编辑
[0102]
64139质粒使用fastdigest bpii酶切,连接入b2msg-f(seq id no:9)和b2msg-r (seq id no:10)引物的退火溶液,构建成sg3质粒,电转入人ips细胞。电转后分别在6小时,12小时,24小时,48小时后进行嘌呤霉素筛选24小时,都无细胞存活。
[0103]
综上所述,本发明的内容并不局限在上述的实施例中,相同领域内的有识之士可以在本发明的技术指导思想之内可以轻易提出其他的实施例,但这种实施例都包括在本发明的范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips