
 商标分类
商标分类  商标转让
商标转让 
取代的烷氧基吡啶基吲哚磺酰胺类的制作方法
 2021-02-02 05:02:51|
2021-02-02 05:02:51| 177|
177| 起点商标网
起点商标网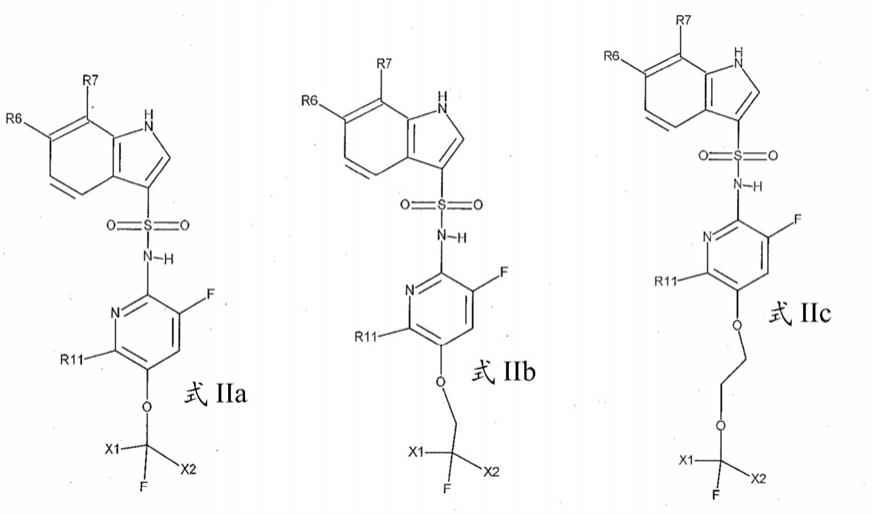
取代的烷氧基吡啶基吲哚磺酰胺类
[0001]
背景
[0002]
g-蛋白偶联受体(gpcr)构成细胞中最大的膜受体家族。它们将细胞外信号转导至细胞内效应系统,并且涉及大量多种生理现象,因此代表了制药药物的最常见靶标,尽管目前的疗法仅靶向一小百分比的gpcr。
[0003]
gpcr响应广泛多种配体。由于人类基因组测序的进展,对于已经鉴定的超过400种gpcr(不包括嗅觉gpcr)中的约25%,仍然缺乏确定的生理学相关配体。这些受体被称为“孤儿gpcr”。预期“脱孤”及其体内作用的鉴定将阐明新的调节机制,并因此公开新的药物靶标。gpr17是否是这样的孤儿受体仍然是一个争论的问题。在系统发育上,gpr17与核苷酸p2y受体和半胱氨酰白三烯(cyslt1,cyslt2)受体密切相关,氨基酸序列同一性分别为约30%至约35%。
[0004]
多组织rna印迹和rt-pcr分析表明,gpr17主要在中枢神经系统(cns)(ciana等人,2006,embo j 25(19):4615;blasius等人,1998,j neurochem 70(4):1357)并且另外在心脏和肾脏即通常经历缺血性损伤的器官中表达。已鉴定出两种人gpr17同种型,它们的n-末端长度不同。短gpr17同种型编码具有典型视紫红质型-7跨膜模体的339个氨基酸-残基蛋白。长同种型编码具有28个氨基酸长的n-末端的受体(blasius等人,1998)。gpr17在脊椎动物物种中是高度保守的(与小鼠和大鼠直向同源物的氨基酸序列约90%同一性),这可能构成在药物发现背景下开发小分子配体和动物模型的有利特征。
[0005]
在最初的脱孤报告中,gpr17被鉴定为尿嘧啶核苷酸和半胱氨酰白三烯(cyslt)ltc4和ltd4的双重受体,分别基于
35
sgtpγs结合和camp抑制试验以及单细胞钙成像(ciana等人,2006,同上)。gpr17功能的证据在不同的细胞背景,例如1321n1,cos7,cho和hek293细胞(ciana等人,2006,同上)中提供。随后,一项独立研究证实了gpr17通过尿嘧啶核苷酸激活,但未能通过cyslt重演激活(benned-jensen和rosenkilde,2010,br j pharmacol,159(5):1092)。然而,最近的独立报告(maekawa等人,2009,pnas 106(28),11685;qi等人,2013,j pharmacol ther 347,1,38;hennen等人,2013,sci signal 6,298)提出在稳定表达gpr17(1321n1,cho,hek293细胞)的不同细胞背景中缺乏对尿嘧啶核苷酸和cyslt的gpr17响应性。还提出了gpr17的新型调节作用:gpr17—与cyslt1受体共表达时—使得cyslt1受体对其内源性脂质介体ltc4和ltd4无响应。需要额外的研究来更深入地探测gpr17的药理学和功能。
[0006]
调节gpr17活性的药物可具有神经保护,抗炎和抗缺血作用,因此可用于治疗脑,心脏和肾缺血和中风(wo2006/045476),和/或用于改善这些事件的恢复(bonfanti等人,cell death and disease,2017,8,e2871)。gpr17调节剂也被认为参与食物摄取,胰岛素和瘦蛋白反应,因此声称其在肥胖症治疗中具有作用(wo2011/113032)。
[0007]
此外,有强有力的证据表明gpr17参与髓鞘化过程,并且负性gpr17调节剂(拮抗剂或反向激动剂)可以作为治疗或缓解髓鞘化障碍如多发性硬化或脊髓损伤的有价值的药物(chen等人,nature neuroscience 2009,12(11):1398-1406;ceruti等人,brain:a journal of neurology 2009 132(pt 8):2206-18;hennen等人,sci signal,6,2013,298;
simon等人j biolchem 291,2016,705;fumagalli等人,neuropharmacology 104,2016,82)。更近来,两组显示,lpc诱导脊髓中(lu等人,scientific reports,2018,8:4502)或胼胝体(ou等人,j.neurosci.,2016,36(41):10560)中脱髓鞘后,成年gpr17-/-敲除小鼠具有快于同窝野生型小鼠的再髓鞘化速率。这再次证实了gpr17在再髓鞘化过程中的潜在关键作用。相反,已经显示gpr17的活化抑制少突神经胶质细胞前体细胞(opc)成熟,从而阻止有效的髓鞘化(simon等人,同上)。因此,有效和选择性的gpr17拮抗剂或反向激动剂的鉴定将在髓鞘化障碍的治疗中具有重要意义。
[0008]
已知几种严重的髓鞘化疾病是由髓鞘化的紊乱引起的,或者是由于髓磷脂的丧失(通常称为脱髓鞘),和/或是由于身体不能适当地形成髓磷脂(有时称为髓鞘化障碍)。髓鞘化疾病可能是特发性疾病或继发于某些触发事件,例如:创伤性脑损伤或病毒感染。髓鞘化疾病可能主要影响中枢神经系统(cns),但也可能与周围神经系统有关。髓鞘化疾病尤其包括多发性硬化,视神经脊髓炎(也称为德维克病(devic
’
s)病),脑白质营养不良,格林-巴利(guillain-barr
é
)综合征和许多其它疾病,如下文进一步详细描述的(还参见例如love,j clin pathol,59,2006,1151,fumagalli等人,同上)。最近神经退行性疾病如阿尔茨海默病,亨廷顿病,帕金森病,肌萎缩侧索硬化(als)和多系统萎缩(msa)也与减少的髓鞘化密切相关(参见例如ettle等人,molneurobiol 53,2016,3046;jellinger和welling,movement disorders,31,2016;1767;kang等人,nature neurosci 6,2013,571;bartzokis,neurochem res(2007)32:1655)。
[0009]
多发性硬化(ms)是一种慢性进行性疾病。它是一种炎性自身免疫疾病,引起少突神经胶质细胞损伤,脱髓鞘和最终的轴突丧失,从而导致严重神经系统疾病的广谱体征和症状,例如,疲劳,头晕,移动和行走问题,言语和吞咽困难,疼痛和其它。ms有多种形式,新症状发生在孤立的发作(复发形式)或随着时间的推移逐渐形成(渐进形式)。虽然某些症状可能在孤立的发作之间完全消失,但严重的神经系统问题通常仍然存在,特别是当疾病前进到更加进行性的形式时。根据美国多发性硬化协会的统计,美国大约有400,000人被诊断为ms,全世界有多达250万人,估计每年在美国诊断出10,000例新病例。多发性硬化在女性中比在男性中更常见,是两到三倍。
[0010]
对于多发性硬化或许多其它髓鞘化疾病,没有已知的因果治疗或治愈方法。治疗通常是症状性的,并且试图通过解决疾病的炎症成分,在发作后改善功能并防止新的发作。这些免疫调节药物通常仅适度有效,特别是如果疾病进展,但可能具有副作用并且耐受性差。此外,大多数可用的药物,如β-干扰素,格拉默醋酸盐或治疗性抗体仅以注射形式提供和/或仅解决疾病的炎症成分而不是直接解决脱髓鞘。其它药物,如皮质类固醇,显示出相当不明显的抗炎和免疫抑制作用,因此可能导致慢性副作用,例如表现为库欣综合征。
[0011]
因此,对用于治疗髓鞘化疾病,如ms的安全有效的药物,优选适合经口服用的药物,存在强烈需求。理想地,这样的药物将通过减少脱髓鞘和/或通过促进受影响的神经元的再髓鞘化来逆转脱髓鞘过程。有效降低gpr17受体活性的化合物可满足这些要求。
[0012]
然而,已知只有很少有效调节gpr17活性的化合物。
[0013]
wo2005/103291提出内源性分子5氨基乙酰丙酸(5-ala)和胆色素原(pbg)作为gpr17的活化配体,公开了gpr17激动剂的镇痛作用,并提出使用gpr17激动剂治疗神经性疼痛和作为gpr17筛选试验的工具。然而,报道的5-ala和pbg的亲和力非常低,并且在测定中
所需的量是显著的,即在5-ala的三位数微摩尔范围内或甚至在pbg的mm范围内,这使得这两种化合物都不适合用于常规筛选试验或甚至用于治疗。此外,pbg是一种化学不稳定的活性化合物,在暴露于空气和光线后迅速分解,使得常规处理变得不切实际。因此,这些化合物不能提供开发治疗有效的负性gpr17调节剂的有希望的起点。
[0014]
孟鲁司特和普仑司特最初是作为白三烯受体拮抗剂开发的,最近发现它们也作用于gpr17受体(ciana等人,embo j.2006,25,4615-4627)。然而,随后的功能测定结果对于孟鲁司特是矛盾的(hennen等人,2013,同上),而用普仑司特对gpr17的药理学抑制促进了原代小鼠(hennen等人,2013,同上)和大鼠(ou等人,j.neurosci.36,2016,10560-10573)少突神经胶质细胞的分化。普仑司特甚至可以显示gpr17抑制在局灶性脱髓鞘的溶血卵磷脂模型中的作用,因为gpr17敲除和普仑司特处理的野生型小鼠显示出较早的再髓鞘化(ou,同上)。这些结果有力地支持了gpr17抑制剂为治疗动物/人类脱髓鞘疾病提供潜力的假设。
[0015]
然而,孟鲁司特和普仑司特对gpr17的亲和力仅在高微摩尔范围内(等人,acs med.chem.lett.2014,5,326-330)。鉴于这两种化合物的高蛋白质结合及其脑渗透性差,它们不太可能达到足够高的游离浓度以适合人类治疗的量结合gpr17受体。此外,由于它们对cyslt1受体具有令人混乱的高度亲和力,因此难以解释用这些化合物在体内获得的结果。us8,623,593公开了某些吲哚-2-甲酸作为gpr17激动剂及其在筛选试验中的用途。然而,这些衍生物全部为有效的激动剂,并且不适合在治疗髓鞘化障碍如ms中所需的下调gpr17活性。此外,这类gpr17激活剂由于其易于电离的羧基而不能充分通过血脑屏障,因此没有适合的先导化合物来产生负性gpr17调节剂。还参见baqi等人,med.chem.commun.,2014,5,86和等人,2014,同上。
[0016]
wo2013/167177提出某些苯基三唑和苯并二氮杂化合物作为gpr17拮抗剂。然而,选择所公开的化合物仅基于计算机模拟筛选结果,并且根本没有提供生物学数据。迄今为止本申请的发明人无法证实该在前专利申请的作者提出的任何所谓配体的gpr17拮抗剂调节活性。因此,对鉴定gpr17的有效调节剂,优选负性调节剂,最优选gpr17的反向激动剂存在需求,它们能够有效降低gpr17活性,优选经口服用时。
[0017]
本发明的描述
[0018]
本发明涉及作为gpr17受体的负性调节剂起作用的化合物。在一个优选的实施方案中,所述化合物充当gpr17受体的负激动剂,因此抑制组成型活性gpr17。
[0019]
本申请的发明人现已发现如本申请中特别定义的在吡啶基对位具有某些氟烷氧基取代基与在“r7”位上具有特定取代基以及在吲哚-吡啶基磺酰胺核心分子的两个其它位置具有至少两个另外取代基的吡啶-2-基-吲哚磺酰胺类提供了非常良好的gpr17抑制活性与改善的特性。例如,如果r7为氢,如本申请中所公开的在“r7”上添加特定取代基有效地减少了cyp 450-1a2诱导作用(其通常与该特定亚类的化合物相关),同时在吡啶基对位上的氟烷氧基取代基提供某些药物动力学优势,例如与其他取代基(包括例如卤素)相比,血浆蛋白结合更少。
[0020]
因此,本发明涉及具有式i的化合物
[0021][0022]
其中
[0023]
r6选自氟、氯、氟甲基和氟甲氧基,
[0024]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基、氟乙基、氟甲氧基和氟乙氧基,
[0025]
r8为氟或甲氧基,
[0026]
r11为氢、氟或甲氧基,
[0027]
l为价键或为选自-ch2-和-ch2-ch2-o-的连接基,
[0028]
x1和x2独立地选自氢和氟,
[0029]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0030]
一个实施方案涉及式i的化合物,其中
[0031]
r6选自氯、氟甲基和氟甲氧基,
[0032]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0033]
r8为氟或甲氧基,
[0034]
r11为氢、氟或甲氧基,
[0035]
l为价键或为选自-ch2-和-ch2-ch2-o-的连接基,
[0036]
x1和x2独立地选自氢和氟,
[0037]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0038]
一个实施方案涉及式i的化合物,其中
[0039]
r6选自氯、一氟甲基、二氟甲基和三氟甲基,优选选自氯和二氟甲基,
[0040]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0041]
r8为氟或甲氧基,
[0042]
r11为氢、氟或甲氧基,
[0043]
l为价键或为选自-ch2-和-ch2-ch2-o-的连接基,
[0044]
x1和x2独立地选自氢和氟,
[0045]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0046]
一个实施方案涉及式i的化合物,其中r6为氯。
[0047]
一个实施方案涉及式i的化合物,其中r6为氟。
[0048]
一个实施方案涉及式i的化合物,其中r6为氟甲基,优选二氟甲基。
[0049]
一个实施方案涉及式i的化合物,其中r7选自氟甲基、环丙基氧基、氟和氯。
[0050]
一个实施方案涉及式i的化合物,其中r7为氟甲基,优选二氟甲基。
[0051]
一个实施方案涉及式i的化合物,其中r7为氟甲氧基,优选二氟甲氧基或三氟甲氧基。
[0052]
一个实施方案涉及式i的化合物,其中r7为氯。
[0053]
一个实施方案涉及式i的化合物,其中r7为氟。
[0054]
一个实施方案涉及式i的化合物,其中r7为环丙基氧基。
[0055]
一个实施方案涉及式i的化合物,其中r7为环丙基。
[0056]
一个实施方案涉及式i的化合物,其中r11为氟或氢。
[0057]
一个实施方案涉及式i的化合物,其中r11为甲氧基。
[0058]
一个实施方案涉及式i的化合物,其中x1为氢。
[0059]
一个实施方案涉及式i的化合物,其中l选自(a)价键,(b)-ch2-,和(c)-ch2-ch2-o-,因此这类化合物具有式ia、ib或ic的结构,其中任何取代基具有如上文对式i所述的含义。
[0060][0061]
一个实施方案涉及具有式ia、ib或ic的化合物,其中r6选自氯和氟甲基,优选选自氯和二氟甲基,
[0062]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0063]
r11选自氢、甲氧基和氟,
[0064]
x1和x2独立地选自氢和氟。
[0065]
一个实施方案涉及如上文所述的式i的化合物,其中r8为氟,且其中l选自(a)价键,(b)-ch2-,和(c)-ch2-ch2-o-,因此这类化合物具有根据式iia、iib或iic的结构,
[0066][0067]
其中所有其它取代基如上所述。
[0068]
一个实施方案涉及式i的化合物,具有式iia、iib或iic之一,其中
[0069]
r6选自氟、氯和氟甲基,优选选自氯和二氟甲基,
[0070]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0071]
r11为氢、甲氧基或氟,优选甲氧基或氟,
[0072]
x1和x2独立地选自氢和氟,其中优选x1为氢,且x2为氟。
[0073]
一个实施方案涉及如上文所述的式i的化合物,其中r8和r11均为氟,且其中l选自(a)价键,(b)-ch2-,和(c)-ch2-ch2-o-,因此这类化合物具有根据式iiia、iiib或iiic的结构,
[0074][0075]
其中所有其它取代基如上所述。
[0076]
一个实施方案涉及式iiia、iiib或iiic的化合物,其中
[0077]
r6选自氟、氯和氟甲基,优选选自氯和二氟甲基,
[0078]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0079]
x1和x2独立地选自氢和氟,
[0080]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0081]
一个实施方案涉及式i的化合物,其中r8为氟,r11为氢,且其中l选自(a)价键,(b)-ch2-,和(c)-ch2-ch2-o-,因此这类化合物具有根据式iva、ivb或ivc的结构,
[0082][0083]
其中所有其它取代基如上所述。
[0084]
一个实施方案涉及式iva、ivb或ivc的化合物,其中
[0085]
r6选自氟、氯和氟甲基,优选选自氯和二氟甲基,
[0086]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0087]
x1和x2独立地选自氢和氟,
[0088]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0089]
一个实施方案涉及如上文所述的式i的化合物,其中r11为甲氧基,且其中l选自(a)价键,(b)-ch2-,和(c)-ch2-ch2-o-,因此这类化合物具有根据式va、vb或vc的结构:
[0090]
[0091]
其中所有其它取代基如上所述。
[0092]
在一个实施方案中,在式va、vb和vc的化合物中,
[0093]
r6选自氟、氯、氟甲基和氟甲氧基,
[0094]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0095]
r8为氟或甲氧基,优选氟,且
[0096]
x1和x2独立地选自氢和氟。
[0097]
在一个实施方案中,在式va、vb和vc的化合物中,
[0098]
r6选自氯、一氟甲基、二氟甲基和三氟甲基,优选选自氯和二氟甲基,
[0099]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0100]
r8为氟或甲氧基,优选氟,
[0101]
x1和x2独立地选自氢和氟,其中在一个实施方案中,x2优选为氢,
[0102]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0103]
在一个优选的实施方案中,在式va、vb和vc的化合物中,
[0104]
r6选自氯、一氟甲基、二氟甲基和三氟甲基,优选选自氯和二氟甲基,
[0105]
r7选自氟、氯、环丙基、环丙基氧基和氟甲基,
[0106]
r8为氟,
[0107]
x1和x2独立地选自氢和氟,其中在一个实施方案中,x2优选为氢,
[0108]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0109]
一个优选的实施方案涉及式i的化合物,其中l为-ch2-,因此这类化合物具有根据式ib的结构,
[0110][0111]
其中
[0112]
r6选自氟、氯、氟甲基和氟甲氧基,
[0113]
r7选自氟、氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0114]
r8为氟或甲氧基,
[0115]
r11为氢、氟或甲氧基,
[0116]
x1和x2独立地选自氢和氟,
[0117]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0118]
一个优选的实施方案涉及式ib的化合物,其中
[0119]
r6选自氯和氟甲基,优选选自氯和二氟甲基,
[0120]
r7选自氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0121]
r8为氟或甲氧基,
[0122]
r11选自氢、甲氧基和氟,
[0123]
x1为氢,且x2为氢或氟。
[0124]
一个优选的实施方案涉及式ib的化合物,其中
[0125]
r6选自氯和氟甲基,优选选自氯和二氟甲基,
[0126]
r7选自氯、环丙基、环丙基氧基、氟甲基和氟甲氧基,
[0127]
r8为氟,
[0128]
r11选自氢和氟,
[0129]
x1为氢,且x2为氢或氟。
[0130]
一个优选的实施方案涉及式ib的化合物,其中
[0131]
r6选自氯和二氟甲基,
[0132]
r7选自氯、环丙基氧基、氟甲基和氟甲氧基,
[0133]
r8为氟,
[0134]
r11选自氢和氟,
[0135]
x1为氢,且x2为氢或氟。
[0136]
一个优选的实施方案涉及式ib的化合物,其中
[0137]
r6选自氟、氯和二氟甲基,
[0138]
r7选自氟、氯、环丙基氧基和氟甲基,
[0139]
r8为氟,
[0140]
r11选自甲氧基和氟,
[0141]
x1为氢,且x2为氢或氟。
[0142]
一个优选的实施方案涉及式ib的化合物,其中
[0143]
r6选自氯和二氟甲基,
[0144]
r7选自氯、环丙基氧基、氟甲基和氟甲氧基,
[0145]
r8为氟,
[0146]
r11为甲氧基,
[0147]
x1为氢,且x2为氢或氟。
[0148]
一个优选的实施方案涉及式ib的化合物,其中
[0149]
r6选自氯和二氟甲基,
[0150]
r7选自氯、环丙基氧基和二氟甲基,
[0151]
r8为氟,
[0152]
r11选自氢、甲氧基和氟,且优选甲氧基,
[0153]
x1为氢,且x2为氢或氟。
[0154]
一个实施方案涉及式ib的化合物,其中
[0155]
r6选自氯和二氟甲基,
[0156]
r7选自氟、氯、环丙基氧基、二氟甲基、二氟甲氧基和三氟甲氧基,
[0157]
r8为甲氧基,
[0158]
r11选自氢和氟,
[0159]
x1为氢,且x2为氢或氟。
[0160]
一个实施方案涉及如上文所述的式ib的化合物,其中r6为二氟甲基。
[0161]
一个实施方案涉及如上文所述的式ib的化合物,其中r7为氯。
[0162]
一个实施方案涉及如上文所述的式ib的化合物,其中r7为氟。
[0163]
一个实施方案涉及如上文所述的式ib的化合物,其中r7为二氟甲基。
[0164]
一个实施方案涉及如上文所述的式ib的化合物,其中r8为氟。
[0165]
一个实施方案涉及如上文所述的式ib的化合物,其中r8为氟,且r11为甲氧基。
[0166]
一个实施方案涉及如上文所述的式ib的化合物,其中r8为氟,且r11为氢。
[0167]
一个实施方案涉及如上文所述的式ib的化合物,其中r8和r11均为氟。
[0168]
一个实施方案涉及如上文所述的式ib的化合物,其中r8为甲氧基。
[0169]
一个实施方案涉及如上文所述的式ib的化合物,其中r8为甲氧基,且r11为氟。
[0170]
一个实施方案涉及如上文所述的式ib的化合物,其中r8为甲氧基,且r11为氢。
[0171]
一个实施方案涉及如上文所述的式ib的化合物,其中r11为氟。
[0172]
一个实施方案涉及如上文所述的式ib的化合物,其中r11为氢。
[0173]
一个实施方案涉及如上文所述的式ib的化合物,其中r11为甲氧基。
[0174]
一个实施方案涉及如上文所述的式ib的化合物,其中x1和x2均为氢。
[0175]
一个实施方案涉及如上文所述的式ib的化合物,其中x1为氢,且x2为氟。
[0176]
一个优选的实施方案涉及式i、ia、ib、ic、iia、iib、iic、iiia、iiib、iiic、iva、ivb、ivc、va、vb或vc的化合物,其中
[0177]
r6为氯,
[0178]
r7选自氟、氯、环丙基氧基、氟甲基和氟甲氧基,且优选选自氯、环丙基氧基和二氟甲基,
[0179]
且其中x1为氢,且x2为氢或氟。
[0180]
一个优选的实施方案涉及式i、ia、ib、ic、iia、iib、iic、iiia、iiib、iiic、iva、ivb、ivc、va、vb或vc的化合物,其中
[0181]
r6为二氟甲基,
[0182]
r7选自氟、氯、环丙基氧基、氟甲基和氟甲氧基,且优选选自氯、环丙基氧基和二氟甲基,
[0183]
且其中x1为氢,且x2为氢或氟。
[0184]
一个实施方案涉及如上文所述的化合物的任意一种,其中x1为氢,且x2为氟。
[0185]
一个实施方案涉及如上文所述的化合物的任意一种,其中x1和x2均为氢。
[0186]
一个优选的实施方案涉及如上文所述的化合物的任意一种,其中r6为氯。
[0187]
一个优选的实施方案涉及如上文所述的化合物的任意一种,其中r6为二氟甲基。
[0188]
一个优选的实施方案涉及如上文所述的化合物的任意一种,其中r7为氯。
[0189]
一个优选的实施方案涉及如上文所述的化合物的任意一种,其中r7为氟。
[0190]
一个优选的实施方案涉及如上文所述的化合物的任意一种,其中r7为二氟甲基。
[0191]
一个实施方案涉及如上文所述的化合物的任意一种,其中r7为一氟甲基。
[0192]
一个实施方案涉及如上文所述的化合物的任意一种,其中r7为三氟甲基。
[0193]
一个实施方案涉及如上文所述的化合物的任意一种,其中r7为环丙基氧基。
[0194]
一个实施方案涉及如上文所述的化合物的任意一种,其中r7为一氟甲氧基。
[0195]
一个实施方案涉及如上文所述的化合物的任意一种,其中r7为二氟甲氧基。
[0196]
一个实施方案涉及如上文所述的化合物的任意一种,其中r7为三氟甲氧基。
[0197]
优选的实施方案涉及选自如下的化合物:
[0198]
6,7-二氯-n-[5-(2,2-二氟乙氧基)-3-甲氧基吡啶-2-基]-1h-吲哚-3-磺酰胺
[0199]
7-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-基]-6-(二氟甲基)-1h-吲哚-3-磺酰胺
[0200]
6-氯-n-[5-[2-(二氟甲氧基)乙氧基]-3-氟-6-甲氧基吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺
[0201]
6-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺
[0202]
n-[5-(2,2-二氟乙氧基)-3-氟吡啶-2-基]-6-(二氟甲基)-7-氟-1h-吲哚-3-磺酰胺
[0203]
6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-7-(三氟甲氧基)-1h-吲哚-3-磺酰胺
[0204]
6-氯-n-[5-(二氟甲氧基)-3-甲氧基吡啶-2-基]-7-(二氟甲基)-1h-吲哚-3-磺酰胺
[0205]
6,7-二氯-n-[5-(二氟甲氧基)-3-甲氧基吡啶-2-基]-1h-吲哚-3-磺酰胺
[0206]
6-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-基]-7-(二氟甲基)-1h-吲哚-3-磺酰胺
[0207]
6-氯-n-[5-(2,2-二氟乙氧基)-3-氟吡啶-2-基]-7-(二氟甲基)-1h-吲哚-3-磺酰胺
[0208]
7-氯-n-[3-氟-5-(2-氟乙氧基)吡啶-2-基]-6-(二氟甲基)-1h-吲哚-3-磺酰胺
[0209]
6-(二氟甲基)-7-氟-n-[3-氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺
[0210]
6-氯-7-(二氟甲基)-n-[3-氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺
[0211]
6,7-二氯-n-[5-(2,2-二氟乙氧基)-3-氟吡啶-2-基]-1h-吲哚-3-磺酰胺
[0212]
6,7-二氯-n-[3-氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺
[0213]
6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺
[0214]
6-氯-n-[5-(2,2-二氟乙氧基)-3,6-二氟吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺
[0215]
6-氯-7-环丙基-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺
[0216]
6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-7-(二氟甲基)-1h-吲哚-3-磺酰胺
[0217]
6-氯-7-环丙基氧基-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺,
[0218]
及其药学上可接受的盐、溶剂合物、同位素和共晶。
[0219]
为了清楚起见,上文描述的化合物的任何定义也包括其任何药学上可接受的盐,
溶剂合物,同位素和共晶。
[0220]
一个实施方案涉及本发明的化合物,其用于疗法中。
[0221]
一个实施方案涉及本发明的化合物,其用于治疗或缓解脱髓鞘疾病。
[0222]
一个实施方案涉及本发明的化合物,其用于治疗或缓解多发性硬化。
[0223]
一个实施方案涉及一种治疗脱髓鞘疾病(包括但不限于多发性硬化)的方法,其包括向有此需要的患者施用治疗有效量的本申请中所定义的化合物。
[0224]
一个实施方案涉及治疗性组合物,其包含至少一种本发明的化合物和药学上可接受的载体。
[0225]
另一个优选的实施方案涉及本发明的化合物,其包含至少一个
18
f同位素,优选在如本申请中公开的化合物之一中所示的氟原子的位置上。作为非限制性实例,在本申请中公开的化合物6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺中,四个氟中的至少一个可以由
18
f同位素表示。这同样适用于本申请中所述的其他含氟的化合物。这些含
18
f的化合物可以优选用作pet示踪剂。
[0226]
另一个优选的实施方案涉及本发明的化合物,其包含至少一个
11
c同位素,优选在如本申请中所示的碳原子的位置上。这些含有
11
c的化合物可以优选用作pet示踪剂。
[0227]
另一个优选的实施方案涉及本发明的化合物,其包含至少一个
123
i、
125
i或
131
i同位素,优选在如本申请中所述的卤素原子的位置上。含
123
i、
125
i或
131
i的化合物可以优选用作spect示踪剂。
[0228]
治疗和诊断应用
[0229]
在一方面,本发明涉及本申请中所述的任何一种化合物,其用于治疗或诊断中,特别是用于治疗动物,特别是人。
[0230]
由于它们的gpr17调节特性,本发明的化合物可以用作药物,并且可以用于治疗和/或预防cns系统的多种疾病。
[0231]
因此,本公开的一个实施方案是如本申请中所述的化合物,其用作药物,特别是用作治疗和/或预防gpr17相关疾病的药物。
[0232]
gpr17相关疾病或障碍为与gpr17信号传导系统功能障碍,如gpr17受体的超表达和/或过度活性有关的疾病。不希望受任何理论的束缚,在某些组织,例如在少突神经胶质细胞祖细胞(opc)中或在少突神经胶质细胞成熟期间,gpr17的活性可能增加、延长或以其它方式改变,这可能是由于激活内源性刺激例如炎性因子。gpr17高活性可能阻止少突神经胶质细胞的分化和有效的髓鞘化,从而促进髓鞘化疾病的出现或进一步发展(参见chen等人,同上)。因此,负性gpr17调节剂可通过降低或关闭gpr17活性并通过支持opc成熟为产生髓磷脂的少突神经胶质细胞来促进髓鞘化(参见例如simon等人,同上)。
[0233]
在一个优选的方面,本发明涉及本申请中所述的任何一种化合物,其用于治疗或诊断中、用于预防或治疗选自髓鞘化障碍和/或与之相关的障碍或综合征,特别是脱髓鞘疾病,例如中枢神经系统的障碍或综合征。在一个实施方案中,本发明的化合物用于在需要的动物中促进、刺激和/或加速再髓鞘化。在一个实施方案中,与本发明化合物的施用有关的再髓鞘化将预防或治疗脱髓鞘疾病,例如但不限于多发性硬化。
[0234]
本发明的化合物还可用于治疗或预防与脑组织损伤有关的障碍综合征、脑血管疾病和某些神经退行性疾病。
[0235]
最近神经退行性疾病与髓鞘化的丧失密切相关。因此,据信保守的少突神经胶质细胞和髓磷脂功能是预防轴突和神经元变性的关键先决条件(参见例如ettle等人,同上)。因此,负性gpr17调节剂可代表对任何与脱髓鞘和/或受影响的髓鞘化相关的神经退行性疾病,例如,als,msa,阿尔茨海默病,亨廷顿病或帕金森病的优异治疗选择。
[0236]
在一个特别优选的方面,本发明的化合物因此可通过口服施用用于预防和/或治疗外周或中枢髓鞘化障碍,特别是中枢神经系统脱髓鞘障碍。在一方面,本发明的化合物通过经口服用用于治疗和/或预防和/或诊断髓鞘化障碍。在一个优选的实施方案中,用本发明化合物治疗的髓鞘化障碍为脱髓鞘障碍。
[0237]
本申请中公开的化合物治疗和/或预防的所述髓鞘化障碍的实例特别是,
[0238]-多发性硬化(ms),包括其各种亚型,
[0239]-视神经脊髓炎(也称为德维克病),
[0240]-慢性复发性炎性视神经炎,急性播散性脑脊髓炎,
[0241]-急性出血性脑白质炎(ahl),
[0242]-脑室周围白质软化,
[0243]-由病毒感染例如由hiv引起的脱髓鞘,或进行性多灶性白质脑病,
[0244]-中央脑桥和脑桥外的髓鞘溶解,
[0245]-由于创伤性脑组织损伤引起的脱髓鞘,包括压迫诱导的脱髓鞘,例如,由肿瘤,
[0246]-响应缺氧、中风或局部缺血或其它心血管疾病的脱髓鞘
[0247]-因暴露于二氧化碳、氰化物或其它cns毒素导致的脱髓鞘,
[0248]-希尔德(schilder)病,
[0249]-巴洛(balo)同心性硬化,
[0250]-围产期脑病,和
[0251]-神经退行性疾病,特别包括
[0252]
ο肌萎缩侧索硬化(als),
[0253]
ο阿尔茨海默病(ad),
[0254]
ο多系统萎缩,
[0255]
ο帕金森病,
[0256]
ο脊髓小脑性共济失调(sca),又称脊髓小脑萎缩,
[0257]
ο亨廷顿病,
[0258]-精神障碍如精神分裂症和双相性精神障碍(参见例如fields,trends neurosci 31,2008,361;tkachev等人,lancet 362,2003,798)。
[0259]-外周髓鞘化疾病,如脑白质营养不良,外周脱髓鞘神经病,德热里纳-索塔斯(dejerine-sottas)综合征或夏科-马里-图思(charcot-marie-tooth)病。
[0260]
cns疾病如脱髓鞘疾病的治疗或预防还包括与该疾病有关的体征和症状的治疗。
[0261]
例如,本发明的化合物用于治疗和/或预防ms的用途还包括治疗和/或预防与ms相关的体征和症状,如对以下的负面影响:视神经(视力丧失,复视),背柱(感觉丧失),皮质脊髓束(痉挛性无力),小脑通路(不协调,构音障碍,眩晕,认知障碍),内侧纵束(侧视凝视复视),脊柱三叉神经束(面部麻木或疼痛),肌肉无力(吞咽受损,膀胱或肠道控制,痉挛),或与潜在疾病如抑郁,焦虑或其它情绪障碍相关的心理影响,全身无力或失眠。
[0262]
因此,本发明的化合物用于治疗髓鞘化疾病,特别是脱髓鞘疾病例如多发性硬化的体征和症状;ms的这类征和症状包括但不限于视力丧失,视力障碍,复视,感觉丧失或损害,虚弱如痉挛性虚弱,运动不协调,眩晕,认知障碍,面部麻木,面部疼痛,吞咽受损,言语受损,膀胱和/或肠道控制受损,痉挛,抑郁,焦虑,情绪障碍,失眠和疲劳。
[0263]
在一个优选的实施方案中,本发明的化合物用于治疗多发性硬化。ms是异质性髓鞘化疾病并且自身可以以各种不同的形式和阶段表现出来,包括但不限于复发-缓解型ms,继发-进展型ms,原发进展型ms,渐进复发型ms,各自取决于活性和疾病进展。因此,在一个实施方案中,如本申请中所述,本发明的化合物用于治疗不同阶段和形式的多发性硬化症。
[0264]
一方面,本发明的化合物用于治疗/预防视神经脊髓炎(也称为德维克病或德维克综合征)。视神经脊髓炎是一种复杂性疾病,其特征在于视神经和脊髓的炎症和脱髓鞘。许多相关症状与ms相似并且包括肌肉无力,特别是肢体肌肉无力,感觉减退和膀胱控制丧失。
[0265]
在一方面,本发明的化合物用于预防和/或治疗als。最近als与少突神经胶质细胞变性和增加的脱髓鞘相关,表明als是负性gpr17调节剂的靶疾病(kang等人,同上;fumagalli等人,neuropharmacology 104,2016,82)。
[0266]
在一方面,本发明的化合物用于预防和/或治疗亨廷顿病。亨廷顿被充分描述为与受影响的髓鞘化相关(bartzokis等人,同上;huang人,neuron 85,2015,1212)。
[0267]
在一方面,本发明的化合物用于预防和/或治疗多系统萎缩。最近msa与脱髓鞘密切相关(ettle,同上,jellinger,同上),提示治疗或预防msa的再髓鞘化策略。
[0268]
在一方面,本发明的化合物用于预防和/或治疗阿尔茨海默病。最近观察到ad与少突神经胶质细胞的细胞死亡增加和局灶性脱髓鞘相关并代表ad中的病理过程(mitew等人,acta neuropathol 119,2010,567)。
[0269]
本发明的一方面涉及通过给予有需要的受试者,包括人患者,治疗有效量的本发明的化合物治疗本申请中所述的任何一种疾病或障碍,特别是髓鞘化疾病如ms,视神经脊髓炎,als,亨廷顿舞蹈病,阿尔茨海默病或其它疾病或障碍的方法。
[0270]
在另一方面,本发明的化合物可以用于预防和治疗脊髓损伤,围产期脑病,中风,局部缺血或脑血管疾病。
[0271]
在一方面,本发明涉及用于预防和/或治疗与髓鞘化障碍相关的综合征或障碍或与脑组织损伤相关的障碍或综合征的方法,其包括对有此需要的患者给予治疗有效量的本申请中所述的化合物。需要这种治疗的患者可以是遭受例如由于机械,化学,病毒或其他创伤导致的脑组织损伤的任何患者。
[0272]
在一方面,本发明涉及用于预防和/或治疗与髓鞘化障碍相关的综合征或障碍,或与中风或其他脑缺血相关的障碍或综合征的方法,该方法包括对有此需要的患者给予治疗有效量的本申请中所述的化合物。有此需要的患者可以是最近经历了脑缺血/中风的任何患者,所述脑缺血/中风例如可能由于栓塞或局部血栓形成而阻塞脑动脉导致。
[0273]
近来,gpr17还与食物摄取、胰岛素控制和肥胖症相关。根据各种报道,gpr17的负性调节剂可能有助于控制食物摄取和治疗肥胖症(参见例如ren等人,diabetes 64,2015;3670)。因此,本发明的一个实施方案涉及本发明的化合物用于预防和/或治疗肥胖症的用途和治疗肥胖症的方法。
[0274]
此外,本发明的化合物可用于治疗或预防其中表达gpr17的组织,例如心脏、肺或
肾。在一个实施方案中,本发明的化合物可以用于治疗或预防肾脏和/或心脏的缺血性疾病。
[0275]
gpr17还与肺部炎症和哮喘有关,例如,它们由屋尘螨诱发(maekawa,j immunol 2010,185(3),1846-1854)。因此,本发明的化合物可以用于治疗哮喘或其他肺部炎症。
[0276]
根据本发明的治疗可包括给予本申请中公开的化合物之一作为cns疾病,特别是髓鞘化疾病或障碍如ms或als的“单独”治疗。或者,本申请中公开的化合物可以在联合疗法中与其它有用的药物一起施用。
[0277]
在一个非限制性实例中,根据本发明的化合物与用于治疗髓鞘化疾病如ms的另一种药物组合,所述另一种药物具有不同的作用模式,例如抗炎药或免疫抑制药。这类化合物包括、但不限于:(i)皮质类固醇,例如泼尼松,甲基泼尼松龙或地塞米松;(ii)β干扰素,例如干扰素β-1a,干扰素β-1b或聚乙二醇干扰素β-1a,(iii)抗-cd20抗体,例如奥瑞珠单抗,利妥昔单抗和奥法木单抗,(iv)格拉默盐,例如乙酸格拉默,(v)富马酸二甲酯,(vi)芬戈莫德和其他鞘氨醇-1-磷酸受体调节剂,例如庞西莫德,辛尼莫德,奥扎尼莫德或拉喹莫德,(vii)二氢乳清酸脱氢酶抑制剂,例如特立氟胺或来氟米特,(viii)抗-整合蛋白α4抗体,例如那他珠单抗,(ix)抗cd52抗体,例如阿仑珠单抗,(x)米托蒽醌,(xi)抗lingo1抗体,例如奥匹单抗(opicinumab),或(xii)其他免疫调节疗法,马赛替尼(masitinib)。
[0278]
同样,如果治疗疼痛性髓鞘化障碍,本发明的化合物可与镇痛药组合。而且,本公开的化合物可以与抗抑郁药组合使用以共同治疗与被治疗的潜在髓鞘化疾病相关的心理影响。
[0279]
在组合疗法中,两种或更多种活性成分可以通过相同的制剂或“部件套装产品”提供,即在单独的盖伦单元中。此外,两种或更多种活性成分,包括本发明的化合物,可以同时或依次给予患者,例如,在间歇疗法中。另外的药物可以通过相同的给药方式或不同的给药方式给药。例如,本发明的gpr17调节剂可以经口服用,而第二种药物可以通过皮下注射给药。
[0280]
在一方面,本发明的化合物可用于诊断和/或监测如本申请进一步描述的gpr17相关疾病,特别是如本申请中所公开的脱髓鞘疾病,优选用于诊断和监测多发性硬化。
[0281]
在一方面,本发明的化合物可用于在体内,例如直接在受试者中,例如使用分子成像技术,或在体外,例如通过检查任何样本如体液或取自受试者的组织,来诊断和/或监测gpr17受体的表达、分布和/或活化。gpr17活性、表达和/或分布的任何此类确定可用于预测、诊断和/或监测(a)如本申请中所述的gpr17相关疾病,特别是髓鞘化疾病,包括但不限于例如,多发性硬化的状态和进展,和(b)与任何这类gpr17相关疾病相关的治疗的功效和/或适用性和/或适当剂量。
[0282]
在一方面,如本申请进一步公开的,本发明的化合物可以用作pet或spect示踪剂,以便进行体内诊断和/或疾病监测。通过它,例如,通过在施用本发明的gpr17 pet或spect示踪剂后对人类患者进行成像,可以在受试者中直接测量gpr17受体的表达、活化和/或分布。这可以促进疾病的正确诊断,可以帮助确定可适用的治疗选择和/或可以用于监测疾病进展和/或监测或预测医学干预的成功,包括治疗药物的选择和适当的施用和/或给药。
[0283]
在一个实施方案中,本发明的pet或spect示踪剂可以与治疗药物联合使用,即作为伴随诊断剂,以便监测和/或预测所述治疗药物在特定受试者中的功效和/或安全性,或
估计药物的适当剂量。
[0284]
一个实施方案涉及本发明的pet或spect示踪剂,其与治疗药物一起用作伴随药物。与本发明的pet或spect示踪剂一起使用的治疗药物可以选自:(a)未标记的本发明的化合物,(b)不同于本发明的化合物的gpr17调节化合物,和(c)用于治疗髓鞘化疾病的药物,包括但不限于用于多发性硬化治疗的药物,其不是gpr17调节剂,如本申请中进一步所述。
[0285]
一个实施方案涉及套装产品,其包含
[0286]
(a)作为第一组分,本发明的pet或spect示踪剂,特别是基于本发明的化合物的pet或pet示踪剂,但其已引入至少一种适用于pet或spect成像的放射性核素,优选选自
18
f,
11
c,
123
i,
125
i和
131
i的放射性核素,
[0287]
(b)作为第二组分,治疗药物,其选自
[0288]
i.本发明的化合物,如本申请进一步定义,且没有引入放射性核素,
[0289]
ii.不同于本发明化合物的gpr17调节化合物,以及iii.用于治疗髓鞘化疾病的药物,包括但不限于用于多发性硬化治疗的药物,但其不具有gpr17调节活性;这类化合物为本领域技术人员已知,包括本申请进一步描述的那些实例。
[0290]
或者,本发明的化合物可用于体外诊断测定法中,例如用于检查受试者的合适体液,如血液,血浆,尿液,唾液或脑脊髓液的任何水平的gpr17表达、活性和/或分布。
[0291]
一个实施方案涉及治疗gpr17相关疾病,特别是髓鞘化疾病,包括但不限于多发性硬化的方法,其中所述方法包括以下步骤:(a)测定受试者的gpr17受体的表达、活性和/或分布,(b)比较所述受试者中gpr17受体的表达、活性和/或分布与一个或多个健康受试者或群体中gpr17受体的表达、活性和/或分布,(c)基于所述受试者与健康受试者或群体的gpr17的表达、活性和/或分布的偏差确定对所述受试者的医学治疗或预防的需要,和(d)通过向所述个体施用治疗药物,该药物适合于治疗gpr17相关疾病或障碍,特别是通过施用gpr17调节剂,优选通过施用一种或多种本发明的化合物,治疗具有gpr17受体的表达、活性和/或分布偏差的受试者。在一个实施方案中,使用本发明的化合物之一,特别是用本发明的pet或spect示踪剂,或通过使用本发明的pet或spect示踪剂体外检查所述受试者德体液或组织,进行gpr17的表达、活性和/或分布的测定(a)。
[0292]
在一个优选的方面,本发明涉及药物组合物,其包含如本申请中所述的化合物和药学上可接受的载体。
[0293]
对于作为医疗药物的给药,可以在包含本公开的化合物和药学上可接受的载体的药物组合物中使用化合物,如本申请进一步定义的。这种药物组合物可以适用于例如经口,静脉内,肌肉内,皮下,鼻,直肠,经颅,颊粘膜或透皮给药,并且可以包含药学上可接受的载体,助剂,稀释剂,稳定剂等。
[0294]
例如,本发明的化合物可以溶解在油,丙二醇或通常用于生产注射剂的其它溶剂中。载体的合适实例包括但不限于生理盐水,聚乙二醇,乙醇,植物油,肉豆蔻酸异丙酯等。可通过溶解,悬浮或乳化在水溶性溶剂(如盐水和5%葡萄糖)或水不溶性溶剂(如植物油,合成脂肪酸甘油酯,高级脂肪酸酯和丙二醇)中,将本发明的化合物配制成注射剂。本发明的制剂可包括任何常规添加剂,例如溶解剂,等渗剂,悬浮剂,乳化剂,稳定剂和防腐剂。
[0295]
在一个实施方案中,本发明的化合物可以经口服用,例如以片剂,胶囊剂,糖锭剂,粉剂,颗粒剂或以液体或半固体形式,包括例如作为非限制性实例的糖浆剂,混悬剂,乳剂
或溶液。
[0296]
口服制剂,可以包含但不限于持续释放剂,崩解剂,填充剂,润滑剂,稳定剂,抗氧化剂,调味剂,分散剂,电解质,缓冲剂,染料或保存剂。适合的赋形剂和制剂是本领域技术人员已知的,并且被公开在标准专著中,例如remington(“the science and practice of pharmacy”,lippincott,williams&wilkins,2000);或被公开在本领域技术人员熟知的其他资源中。
[0297]
片剂可以例如通过如下步骤来制备:将至少一种本发明的化合物与至少一种无毒的药学上可接受的赋形剂如粘合剂,填充剂/稀释剂,崩解剂,增塑剂等和任选的溶剂(水性或非水性)混合,然后通过包括但不限于干压缩,干法制粒,湿法制粒,喷雾干燥或熔融挤出的方法将混合物加工成片剂。片剂可以是未包衣的,或通过已知技术包衣,以掩盖令人不愉快的味道药物的不良味道,或延迟活性成分在胃肠道中的崩解和吸收。片剂可以提供本发明化合物的即释或缓释。
[0298]
典型的缓释剂为例如在与水接触时溶胀的那些,例如聚乙烯吡咯烷酮,羟乙基纤维素,羟丙基纤维素,其他纤维素醚,淀粉,预胶化淀粉,聚甲基丙烯酸酯,聚醋酸乙烯酯,微晶纤维素,葡聚糖和它们的混合物。崩解剂的非限制性实例包括预糊化淀粉,淀粉羟乙酸钠,微晶纤维素,羧甲基纤维素钠(cmc-na),交联cmc-na和低取代的羟丙基纤维素,以及它们的混合物。适合的填充剂和粘合剂包括但不限于微晶纤维素,粉状纤维素,乳糖(无水或一水合物),可压缩糖,淀粉(例如玉米淀粉或马铃薯淀粉),预胶化淀粉,果糖,蔗糖,右旋糖,葡聚糖,其他糖类,例如甘露糖糖醇,麦芽糖醇,山梨醇,乳糖醇和蔗糖,硅化微晶纤维素,磷酸氢钙,磷酸氢钙二水合物,磷酸二钙二水合物,磷酸三钙,乳酸钙或它们的混合物。润滑剂、防粘剂和/或助流剂包括硬脂酸,硬脂酸镁,硬脂酸钙,月桂基硫酸钠,氢化植物油,氢化蓖麻油,硬脂酰富马酸钠,聚乙二醇,山萮酸甘油酯,滑石粉,玉米淀粉,二氧化硅等,包括混合物。
[0299]
也可配制本发明的化合物用于通过注射,例如通过推注或输注经肠胃外给药。可以提供用于注射的组合物,其随时可用,并且可以采用诸如油性或水性介质中的悬浮液,溶液或乳液的形式,并且可以含有赋形剂,例如悬浮剂,稳定剂,防腐剂和/或分散剂。或者,活性成分可以是粉末形式,用于在使用前在与适合的介质例如无菌无热原水或生理盐水构建。
[0300]
对于鼻腔给药或通过吸入给药,本发明的化合物可以以气溶胶喷雾剂的形式方便地以加压包装或喷雾器的形式递送,使用适合的推进剂,例如二氯二氟甲烷,氟三氯甲烷,二氯四氟乙烷,二氧化碳或其它适合的气体或气体混合物。
[0301]
对于眼科给药,本发明中使用的化合物可以方便地配制成等渗的,ph调节的无菌盐的微粉化悬浮液,含有或不含防腐剂如杀菌剂或杀真菌剂,例如硝酸苯汞,氯化苄基氯化铵或醋酸氯己定。或者,对于眼科给药,化合物可以配制成软膏如凡士林。
[0302]
对于直肠给药,用于本发明的化合物可以方便地配制成栓剂。这些可以通过将活性组分与适合的无刺激性赋形剂混合来制备,所述赋形剂在室温是固体但在直肠温度下是液体,因此将在直肠中熔化以释放活性组分。这些材料包括例如可可脂,蜂蜡和聚乙二醇。
[0303]
在一个实施方案中,化合物可以透皮给药。这种给药方式防止所谓的经口服用的首过效应,而且允许提供更恒定的血浆水平,这在某些情况下是特别有利的。透皮形式的设
计,例如软膏或乳膏或其它透皮系统,例如,贴片或电泳装置通常是本领域已知的,参见例如venkatraman和gale,biomaterials 1998,vol 19,p1119;prausnitz和langer,nat biotechnology 2008,vol 26.11p1261;wo2001/47503;wo2009/000262;wo99/49852;wo 07/094876。
[0304]
根据本发明的化合物的可优选剂量水平取决于多种因素,包括患者的状况和体重,特定疾病的严重程度,剂型,给药途径和期限,但可以由本领域技术人员适当地选择。在各种实施方案中,化合物的施用量为每天0.001至10毫克/千克体重,或每天0.03至1毫克/千克体重。个体剂量可以为每天约0.1至1000毫克活性成分,约0.2至750毫克/天,约0.3至500毫克/天,0.5至300毫克/天,或1至100毫克/天。剂量可以一天一次施用,或一天几次施用,每次分开给药。
[0305]
本发明的另一方面是套装产品,其包含如本申请中所述的药物或药物组合物,及其使用说明。
[0306]
定义
[0307]
本发明的化合物包括如本申请进一步描述的式i,ia,ib,ic,iia,iib,iic,iiia,iiib,iiic,iva,ivb和ivc所涵盖的化合物,以及在本说明书和实验部分中公开的最终个体化合物。
[0308]
除非另有明确说明,否则对根据本发明的化合物的任何提及还包括这些化合物的药学上可接受的盐,溶剂合物,同位素和共晶。
[0309]
术语“药学上可接受的盐”涉及所述化合物可以形成并且根据本发明适合于对受试者,特别是人受试者给药的任何盐。这些盐包括但不限于酸加成盐,其与无机酸如盐酸,氢溴酸,硫酸,硝酸,磷酸等形成,或与有机酸如乙酸,丙酸,己酸,环戊烷丙酸,乙醇酸,丙酮酸,乳酸,丙二酸,琥珀酸,苹果酸,马来酸,富马酸,酒石酸,柠檬酸,苯甲酸,3-(4-羟基苯甲酰基)苯甲酸,肉桂酸,扁桃酸,甲磺酸,乙磺酸,1,2-乙烷-二磺酸,2-羟基乙磺酸,苯磺酸,4-氯苯磺酸,2-萘磺酸,4-甲苯磺酸,樟脑磺酸,4-甲基二环[2.2.2]辛-2-烯-1-甲酸,葡庚糖酸,3-苯基丙酸,三甲基乙酸,叔丁基乙酸,月桂基硫酸,葡糖酸,谷氨酸,羟基萘甲酸,水杨酸,硬脂酸和粘康酸等形成。其它盐包括2,2-二氯乙酸盐,己二酸盐,藻酸盐,抗坏血酸盐,天冬氨酸盐,2-乙酰氨基苯甲酸盐,己酸盐,癸酸盐,樟脑酸盐,环己基氨基磺酸盐,月桂基硫酸盐,乙二酸盐,乙磺酸盐,羟乙基磺酸盐,甲酸盐,龙胆酸盐,龙胆酸盐,葡萄糖酸盐,葡萄糖醛酸盐,氧代戊二酸盐,马尿酸盐,乳糖酸盐,萘二磺酸盐,昔萘酸盐,烟酸盐,油酸盐,乳清酸盐,草酸盐,棕榈酸盐,双羟萘酸盐,吡酮酸盐,对氨基水杨酸盐,癸二酸盐,丹宁酸盐,硫氰酸盐,十一碳烯酸盐等;或当母体化合物中存在的酸性质子被替代时形成的盐,如被氨,精氨酸,苯乙苄胺,n,n-二苄基乙二胺,钙,胆碱,癸醇,二乙醇胺,二乙胺,乙醇胺,乙二胺,葡甲胺,甘氨酸,海巴明,咪唑,赖氨酸,镁,羟乙基吗啉,哌嗪,钾,吡咯乙醇,钠,三乙醇胺,氨丁三醇或锌替代时形成的盐。
[0310]
本发明在其范围内包括本申请定义的化合物的溶剂合物。“溶剂合物”是由活性化合物和第二组分(溶剂)形成的晶体,其以分离的形式在室温为液体。这些溶剂合物可以用普通的有机溶剂形成,例如,烃类溶剂,如苯或甲苯;氯化溶剂,如氯仿或二氯甲烷;醇类溶剂,如甲醇,乙醇或异丙醇;醚类溶剂,如乙醚或四氢呋喃;或酯类溶剂,如乙酸乙酯。或者,本申请化合物的溶剂合物可以用水形成,在这种情况下它们是水合物。
[0311]
本发明在其范围内还包括共晶。术语“共晶”用于描述中性分子组分以确定的化学计量比存在于结晶化合物中的情况。药物共晶的制备使得能够对活性药物成分的结晶形式进行修饰,这反过来可以改变其物理化学性质而不损害其预期的生物活性。可以与活性药物成分一起存在于共晶中的共晶形成剂的实例包括l-抗坏血酸,柠檬酸,戊二酸,肉桂酸,扁桃酸,尿素和烟酰胺。
[0312]
本发明还包括本发明的化合物的所有适合的同位素变体。本发明的化合物的“同位素变体”或简称为“同位素”定义为其中至少一个原子被具有相同原子序数但原子质量不同于通常在自然界中发现的原子质量的原子取代的变体,其中最丰富的同位素是优选的。可以结合到本发明的化合物中的同位素的实例分别包括氢,碳,氮,氧,硫,氟和氯的同位素,例如2h,3h,
11
c,
13
c,
14
c,
15
n,
17
o,
18
o,
35
s,
18
f和
36
cl。本发明的某些同位素变体,例如其中掺入放射性同位素如3h或
14
c的那些,可用于药物和/或底物组织分布研究。氚(即3h)和碳-14(即
14
c)同位素因其易于制备和可检测性而特别优选。此外,用诸如氘的同位素(即2h)取代可以因更大的代谢稳定性而提供某些治疗优势,例如,体内半衰期延长,剂量需求减少,因此在某些情况下可能是优选的。本发明的化合物的同位素变体通常可以通过常规方法使用合适试剂的适当同位素变体来制备。
[0313]
其中至少一个原子被相同或不同原子的放射性同位素(放射性同位素)替代的那些化合物也是本发明的组成部分,所述化合物可用于体内成像技术,如单光子发射计算机体层摄影术(spect)或正电子发射体层摄影术(pet)。
[0314]
可用于spect研究的此类gpr17调节剂的同位素变化形式的实例(本申请中此类化合物称为“spect示踪剂”)是其中引入
99m
tc,
111
in,
82
rb,
137
cs,
123
i,
125
i,
131
i,
67
ga,
192
ir或
201
tl,优选
123
i的化合物。例如,为了将本发明的化合物用作spect示踪剂,可以将
123
i同位素引入如本申请中所公开的gpr17调节剂中。作为非限制性实例,为了将化合物用作spect示踪剂,可以将选自
123
i,
125
i和
131
i的放射性核素引入本发明的化合物中。在一个实施方案中,本发明的spect示踪剂可以基于本申请中公开的含卤素的gpr17调节剂的结构,其中放射性核素
123
i,
125
i和
131
i之一已被引入卤素,优选碘原子的位置。
[0315]
因此,术语“本发明的spect示踪剂”涉及本专利申请中所述并且具有如本申请进一步定义的本发明化合物的任意一种的结构的化合物,其中已经引入了至少一种适合于spect成像的放射性同位素。这包括但不限于
99m
tc,
111
in,
82
rb,
137
cs,
123
i,
125
i,
131
i,
67
ga,
192
ir或
201
tl。
[0316]
可用于pet应用的gpr17调节剂衍生物(本申请中称为“pet示踪剂”)的实例为其中引入了
11
c,
13
n,
15
o,
18
f,
76
br或
124
i的化合物。例如,为了将化合物用作pet示踪剂,可以将
18
f同位素引入本发明的化合物中。在一个实施方案中,pet示踪剂可以基于本申请中公开的含氟gpr17调节剂的结构,其中相应的放射性核素
18
f已被引入氟原子的位置。这同样适用于分别引入至少一个
11
c,
13
n,
15
o,
76
br或
124
i,而不是“未标记的”碳,氮,氧,溴或碘原子(参见例如,pimlott和sutherland,chem soc rev 2011,40,149;van der born等人,chem soc rev 2017,46,4709)。
[0317]
因此,术语“本发明的pet示踪剂”涉及本申请进一步定义的本发明的化合物,其中已经引入至少一个放射性同位素,其适合于pet成像。它包括、但不限于
11
c、
13
n、
15
o、
18
f、
76
br或
124
i。
[0318]
本发明在其范围内包括本发明化合物的前药。通常,这样的前药为本申请所述化合物的功能性衍生物,其在体内易于转化,例如通过肠道或血液中的内源性酶转化为本申请中所述的所需gpr17调节化合物。选择和制备适合的前药衍生物的常规方法描述于,例如design of prodrugs,ed.h。bundgaard,elsevier,1985。
[0319]
取决于其取代模式,本发明的化合物可以具有或不具有一个或多个光学立体中心,并且可以或不以不同的对映异构体或非对映异构体存在。任何这样的对映异构体,非对映异构体或其它光学异构体都包括在本发明的范围内。
[0320]
本发明的化合物也可以以不同的晶型存在,即作为多晶型物,所有这些都包括在本发明中。
[0321]
本发明的化合物可以包含在药物组合物中,该药物组合物还可以包含药学上可接受的载体。“药学上可接受的载体”是指稀释剂,助剂,赋形剂或载体,或与本发明的化合物一起施用的其它成分,并且本领域技术人员将理解为药学上可接受的。
[0322]
如本申请中所述,本发明的化合物可用于预防和/或治疗动物,特别是人类的某些疾病或障碍。
[0323]“预防”(preventing)或“预防”(prevention)是指降低获得疾病或障碍的风险(即,导致疾病的至少一种临床症状不在可能暴露于所述疾病或易患所述疾病但尚未经历或表现出疾病的症状的受试者,特别是人类受试者中发生)。
[0324]
在一个实施方案中,“治疗”(treating)或“治疗”(treatment)任何疾病或障碍包括改善疾病或障碍(即,阻止或减少疾病的发展或至少减少疾病的临床症状之一)。在另一个实施方案中,“治疗”或“治疗”是指改善至少一种物理参数,其可以是或可以不是受试者,特别是人受试者可辨别的,但是基于待治疗的疾病或障碍或与其相关。在另一个实施方案中,“治疗”或“治疗”是指物理上(例如,在不可辨别的症状上可辨别的稳定性),生理学上(例如,生理参数的稳定化)调节或缓解疾病或障碍或两者。在另一个实施方案中,“治疗”或“治疗”是指延迟疾病或障碍的发作或进展。因此,“治疗”或“治疗”包括对潜在疾病或障碍的任何因果治疗(即疾病改变),以及疾病或障碍的体征和症状的任何治疗(无论是否有疾病改变),以及任何疾病或障碍或其体征和症状的减轻或改善。
[0325]
在一个实施方案中,疾病或障碍的“诊断”(diagnosis),“诊断”(diagnoses)或“诊断”(diagnosing)包括鉴定和测量与所述疾病相关的体征和症状。“诊断”,“诊断”或“诊断”包括但不限于检测和/或测量作为gpr17相关疾病或障碍指示的gpr17受体与健康受试者相比的减少,增加或不正确(例如,时间或地点)的表达,激活或分布。在一个实例中,gpr17配体可以以pet或spect示踪剂的形式用于这种诊断,包括髓鞘化疾病的诊断。
[0326]
术语“疾病”和“障碍”在本申请中基本上可互换使用。
[0327]“监测”是指在一段时间期限内观察疾病、障碍或至少一种医学参数。“监测”还包括在“伴随药物”的帮助下观察治疗药物的作用。
[0328]
如本申请所用的“伴随诊断剂”是指可以与治疗药物联合使用的化合物,目的是确定所述治疗药物对特定患者的适用性(例如,在安全性和功效方面)。“伴随诊断剂”的使用可包括诊断和监测步骤。
[0329]
术语“动物”和“受试者”包括人。术语“人”,“患者”和“人类受试者”在本申请中典型地可互换使用,有明确指示的除外。
[0330]
本发明还涉及治疗如本申请更详细描述的动物疾病或障碍,特别是人类疾病或障碍的方法,其包括以治疗有效量给予本发明的化合物。
[0331]“治疗有效量”是指当被给予受试者,特别是人受试者用于治疗疾病时,足以实现对该疾病的这种治疗的化合物的量。“治疗有效量”可以根据化合物,疾病及其严重程度,以及被治疗的受试者,特别是人类受试者的状况,年龄,体重,性别等而变化。
[0332]
如本申请所用的术语“多发性硬化(症)”是指在2018american版icd-10-cm诊断代码的第g35部分中分类的疾病。
[0333]
如本申请所用的术语“gpr17调节剂”意在描述能够调节gpr17受体活性的化合物,特别是能够降低gpr17活性的化合物。这种“负性gpr17调节剂”包括能够阻断gpr17激动剂作用的gpr17拮抗剂,以及也能够抑制组成型活性gpr17受体或受体变体的gpr17反向激动剂。本发明优选的gpr17调节剂为反向gpr17激动剂。
[0334]
所使用的术语“氟甲基”是指被一个(“一氟甲基”)、两个(“二氟甲基”)或三个(“三氟甲基”)氟原子取代的甲基。特别优选的氟烷基为二氟甲基:-chf2。
[0335]
所使用的术语“氟甲氧基”是指被一个(“一氟甲氧基”)、两个(“二氟甲氧基”)或三个(“三氟甲氧基”)氟原子取代的甲氧基。氟烷氧基的实例为三氟甲氧基-ochf3。
[0336]
本申请所使用的术语“环丙基”是指衍生自具有三个成环碳原子的环状饱和烃的单价基团,其可以是未取代的或被一个或多个本申请进一步所示的取代基取代。
[0337]
实验部分:
[0338]
a.化学
[0339]
下面更详细地描述本发明的化合物及其合成途径。
[0340]
a-i制备所述化合物的通用方法
[0341]
如合成有机化学领域的技术人员所理解的,可以按照与常规方法类似的方式制备根据本发明的式i的化合物。
[0342]
a-i制备所述化合物的通用方法
[0343]
如合成有机化学领域的技术人员所理解的,可以按照与常规方法类似的方式制备根据本发明的式i的化合物。
[0344]
任何涉及本申请中的通式i的化合物的合成同样适用于子通式ia、ib、ic、iia、iib、iic、iiia、iiib、iiic、iva、ivb和ivc的适用化合物以及本申请中公开的具体实施例的化合物。
[0345]
根据一个实施方案,可以根据如下反应通过使式xii的磺酰氯与式x的苯胺反应,制备通式i的一些化合物:
[0346][0347]
该反应可以在室温在碱例如用作溶剂的吡啶存在下进行,或者通过在60-100℃范围内的温度加热,可能在催化量的二甲基氨基吡啶存在下进行。
[0348]
或者,某些通式i的化合物可以根据以下反应式通过对式i-p的化合物进行原位脱保护来制备,其中p为保护基,例如苯基磺酰基(phso2):
[0349][0350]
式i-p化合物可以通过使式xii-p的磺酰胺氯与式x的苯胺反应来制备。该反应可以在碱例如用作溶剂的吡啶存在下在室温或通过在60-100℃范围内的温度加热而进行。
[0351]
某些式x的化合物可以通过将式x-p的化合物脱保护来制备,其中p为保护基,例如对-甲氧基苄基(pmb),使用酸性条件,例如纯三氟乙酸,或使用溶剂,例如二氯甲烷。其他脱保护条件也可以为在催化剂如钯或氢氧化钯(可能在碳上活化)存在下使用还原剂如氢在溶剂如甲醇中。
[0352][0353]
某些式x-p的化合物可以通过使式x-oh的苯胺与式y-lg的分子反应来制备,其中lg为离去基团,例如溴,在例如k2co3或koh这样的碱存在下于极性溶剂,例如dmf或acn中。
[0354][0355]
或者,可以在活化剂或混合物如氰基甲基亚甲基三丁基磷杂环己烷(cmbp)或三苯膦和偶氮二羧酸二异丙酯(diad)存在下,通过将式x-oh的苯胺与式y-oh的分子在60℃-120℃范围内的温度在溶剂如甲苯或thf中加热,进行mitsunobu-型反应,制备某些式x-p的化合物。
[0356][0357]
式x-oh的苯胺可以根据本领域技术人员已知的任何方法或使用文献中描述的方法来制备。
[0358]
式xii的化合物可以根据如下反应式通过式xi的化合物的氯磺酰化来制备:
[0359]
[0360]
该反应可以在磺酰化剂例如氯磺酸存在下在极性溶剂例如乙腈中在室温进行。
[0361]
类似地,式xii-p的化合物,其中p为保护基,例如苯磺酰基,可以根据如下反应式通过式xi-p的化合物的氯磺酰化来制备:
[0362][0363]
该反应可以在氯磺酸存在下在极性溶剂如乙腈中在室温进行。
[0364]
式xi-p的化合物,其中p为保护基,例如苯磺酰基,可以根据以下反应式通过保护式xi的化合物来制备:
[0365][0366]
该反应可以根据本领域技术人员已知的任何方法进行。
[0367]
式xi的化合物可以通过本领域技术人员熟知的适合方法来制备。
[0368]
或者,某些式xi的化合物可以根据以下反应式,通过邻位取代的硝基芳烃xii与乙烯基格氏试剂xiii的反应(bartoli吲哚合成)来制备:
[0369][0370]
该反应可以在-20℃至-78℃范围内的低温,在极性溶剂如四氢呋喃中使用乙烯基格氏试剂如溴化乙烯基镁进行。
[0371]
或者,可以使用文献中描述的或本领域技术人员已知的方法,通过在已经组装的具有式i的化合物的类似物上进行官能团转化,制备具有通式i的一些化合物。
[0372]
特别地,一些式i化合物(其中r7为环丙基)可以从式i化合物(其中r7为卤原子,优选为溴)开始,根据本领域技术人员已知的方法,在相应的烃基代硼酸,钯盐,例如乙酸钯,膦,例如三环己基膦和碱例如磷酸三钾存在下,在溶剂例如甲苯中通过suzuki-型偶联进行制备。
[0373]
a-ii.缩写/重现的试剂
[0374]
ac:乙酰基
[0375]
acn:乙腈
[0376]
盐水:饱和氯化钠水溶液
[0377]
nbu:正丁基
[0378]
tbu:叔丁基
[0379]
cmbp:氰基亚甲基三丁基膦
[0380]
cy:环己基
[0381]
dast:氟化二乙基氨基硫
[0382]
dcm:二氯甲烷
[0383]
diad:偶氮二羧酸二异丙酯
[0384]
dmac:n,n-二甲基乙酰胺
[0385]
dmap:4-二甲基氨基吡啶
[0386]
dmf:n,n-二甲基甲酰胺
[0387]
dmso:二甲亚砜
[0388]
dppf:1,1'-双(二苯基膦基)二茂铁
[0389]
es
+
:电喷雾正离子化
[0390]
es-:电喷雾负离子化
[0391]
esi:电喷雾离子化
[0392]
etoac:乙酸乙酯
[0393]
h:小时
[0394]
lc:液相色谱法
[0395]
lcms:液相色谱质谱法
[0396]
me:甲基
[0397]
meoh:甲醇
[0398]
min:分钟
[0399]
mw:微波炉
[0400]
nbs:n-溴代琥珀酰亚胺
[0401]
nmr:核磁共振
[0402]
pin:频哪醇合(pinacolato)
[0403]
pmb:对甲氧基苄基
[0404]
rt:室温
[0405]
tbahsa:酸式硫酸四丁基铵
[0406]
tfa:三氟乙酸
[0407]
thf:四氢呋喃
[0408]
tlc:薄层色谱法
[0409]
a-iii.分析方法
[0410]
通常使用商业溶剂和试剂,没有进一步纯化,适当时包括无水溶剂(通常为来自aldrich chemical company的sure-seal
tm
产品或来自acros organics的acroseal
tm
)。通常,反应后接着进行薄层色谱法或液相色谱质谱法分析。
[0411]
使用如下不同的方法和仪器以lcms模式进行质谱测量:
[0412]-碱性lcms方法1:
[0413]
qda waters单四极杆质谱仪用于lcms分析。该光谱仪配有esi源和具有二极管阵
列检测器(200至400nm)的uplc acquity hclass。数据以完全ms扫描从m/z 70至800以具有碱性洗脱的正/负模式获得。反相分离在45℃在用于碱性洗脱的waters acquity uplc beh c18 1.7μm(2.1
×
50mm)柱上进行。根据表1用水/acn/甲酸铵(95/5/63mg/l)(溶剂a)和acn/水/甲酸铵(95/5/63mg/l)(溶剂b)进行梯度洗脱。注射体积:1μl。ms中全流量。
[0414]
表1:
[0415]
时间(min)a(%)b(%)流速(ml/min)09910.40.39910.43.201000.43.2501000.5401000.54.19910.44.89910.4
[0416]-碱性lcms方法2:
[0417]
质谱(ms)谱图记录在具有电喷雾离子化(esi)的lcms-2010ev质谱仪(shimadzu)上,其与使用xbridge c18-2.1x30mm,2.5μm(waters)柱的hplc modular prominence(shimadzu)偶联。注射体积为3μl的样品溶液,其浓度为约1mg/ml。碱性条件的流动相是a)5mm甲酸铵+0.1%氨水溶液b)5%流动相a+0.1%氨的乙腈溶液的混合物。使用的梯度如下-在4min内5:95(b/a)至95:5(b/a)并且在接下来1min保持95:5(b/a)。
[0418]-中性lcms方法3:
[0419]
使用以下程序在lcms仪器(applied biosystems api 2000lc/ms/ms,hplc agilent 1100)上记录质谱(ms)谱图:将所述化合物以1.0mg ml-1
的浓度溶解在acn(溶剂a)或水(含有2mm乙酸铵):meoh 90:10(溶剂b)中,并且如果需要超声处理直至完全溶解。然后,将10μl溶液注入phenomenex luna c18 hplc柱(50
×
2.00mm,粒径3μm),并用以下梯度进行洗脱:水:acn(梯度a)或水:meoh(梯度b)在10min内从90:10到0:100,1min后开始梯度,然后在纯有机溶剂中以300μl min-1
的流速洗脱10min。使用二极管阵列检测器(dad)从220至400nm检测uv吸收。
[0420]-酸性lcms方法4:
[0421]
在偶联至uv检测(230-400nm和215nm)和mass spec detection agilent 6120质谱仪(es)m/z 120-800的agilent 1200-6120lc-ms系统上,使用x-bridge c18 waters 2.1x 20mm,2.5μm柱进行hplc-ms。使用表2中所示的流动相a(10mm甲酸铵水溶液+0.1%甲酸)和流动相b(乙腈+5%水+0.1%甲酸)梯度,以1ml/min的流速进行洗脱。
[0422]
表2:
[0423]
时间(min)a(%)b(%)09461.55952.255952.50946
[0424]
可以通过正相色谱法,(酸性或碱性)反相色谱法或重结晶纯化粗制物质。
[0425]
使用硅胶柱(100:200目硅胶或用于急骤色谱系统的柱,例如或teledyne isco的isolera
tm four)进行正相色谱法。
[0426]
使用两种不同的仪器并根据以下方法进行制备型反相色谱法:
[0427]-碱性制备型lcms方法1:
[0428]
lcms纯化使用sqd或qm waters单四极杆质谱仪进行ms检测。该光谱仪配有esi源、waters 2525二元泵,其连接有2767样品管理器和二极管阵列检测器(210-400nm)。
[0429]
ms参数:esi毛细管电压3kv。锥孔和提取锥孔电压10。源部件温度120℃。去溶剂化温度为300℃。锥孔气体流量30l/h(氮气),去溶剂化气体流量650l/h。数据以m/z 100至850的全ms扫描以正/负模式获得。
[0430]
lc参数:反相分离在室温在xbridge制备型obd c18柱(5μm,30
×
50mm)上进行。用溶剂a1(h2o+nh4hco
3 10mm+50μl/l nh4oh)和溶剂b1(100%acn)(ph~8.5)进行梯度洗脱。hplc流速:35ml/min至45ml/min,注射体积:990μl。分流比设定为+/-1/6000至ms(表3)。
[0431]
表3:
[0432]
时间(min)a1(%)b1(%)流速(ml/min)09553519553571090357.5595359595359.1595451259545
[0433]-中性rp-hplc方法2:
[0434]
使用rp-hplc柱(knauer 20mm i.d.,eurospher-100c18)在knauer smartline 1050hplc系统上进行最终产物的hplc纯化。将产物溶解在甲醇(20mg/8ml)中,并进行反相hplc,应用甲醇/水(70:30至100:0,历时24min)的梯度。
[0435]
nmr光谱记录在不同的仪器上:
[0436]-bruker avanceiii 400mhz-ultrashield nmr光谱仪,其配备有运行topspin 3.2软件的windows 7professional工作站和5mm双重共振宽带探针(pabbi 1h/19f-bb z-grd z82021/0075)或1mm三重共振探针(patxi 1h/d-13c/15n z-grd z868301/004)。
[0437]-varian 400mhz nmr光谱仪,采集时间(at)=2.0秒,弛豫延迟(d1)=2.0秒和线加宽(lb)=0.5hz。
[0438]-bruker avance drx 500mhz nmr光谱仪
[0439]-bruker avance iii 600mhz nmr光谱仪
[0440]
化学位移参考源自氘代溶剂(dmso-d6,苯-d6或cdcl3)的残余质子的信号。化学位移以百万分率(ppm)和以赫兹(hz)表示的偶合常数(j)给出。以宽峰(br),单重峰(s),双重峰(d),三重峰(t),四重峰(q)和多重峰(m)给出自旋多重性。
[0441]
通常在最终分析并提交至生物测试之前在真空下干燥产品。
[0442]
a-iv:实施例化合物和合成
[0443]
以下化合物的名称是iupac名称,对于式x,xi,xii的中间体由biovia draw 16.1版本产生,对于式i的实施例化合物由pipeline pilot 2018,应用openeye oemetachem 1.4.5版本产生。
[0444]
中间体
[0445]
当可商购时,原料根据其cas注册号鉴定。
[0446]
a.式x的中间体的合成
[0447]
a.1. 3,6-二氟-5-(2-氟乙氧基)吡啶-2-胺x-1的合成:
[0448][0449]
步骤-1:5-氯-3,6-二氟-吡啶-2-胺x-1a的合成
[0450]
向3-氯-2,5,6-三氟-吡啶(0.50g,2.98mmol)在dmso(10ml)中的溶液中加入25%nh3水溶液(4ml),将该反应混合物在钢桶中在100℃加热12h。通过tlc和lcms监测反应进程。完成后,用h2o(200ml)稀释该反应混合物,用etoac(400ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到5-氯-3,6-二氟-吡啶-2-胺x-1a(0.41g粗制品),为黄色固体。
[0451]
将该化合物原样用于下一步反应,未经进一步纯化。
[0452]1h nmr(400mhz,dmso-d6)δ6.85(s,2h),7.80-7.85(m,1h)。
[0453]
步骤-2:3,6-二氟吡啶-2-胺x-1b的合成
[0454]
向5-氯-3,6-二氟-吡啶-2-胺x-1a(0.50g,3.03mmol)在meoh(100ml)中的溶液中
加入三乙胺(5ml)和pd/c(0.40g),将该反应混合物在室温在氢气压力气氛中在帕尔摇瓶中搅拌10h。通过tlc和lcms监测反应进程。完成后,通过硅藻土过滤该反应混合物,真空浓缩滤液。残余物用h2o(200ml)稀释,用10%meoh的dcm溶液(200ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到3,6-二氟吡啶-2-胺x-1b(0.21g粗制品),为灰白色固体。
[0455]
将该化合物原样用于下一步反应,未经进一步纯化。
[0456]
碱性lc-ms方法2(es
+
):130(m)
+
,91%纯度。
[0457]1h nmr(400mhz,dmso-d6)δ6.09-6.11(m,1h)6.57(brs,2h)7.44-7.50(m,1h)。
[0458]
步骤-3:5-溴-3,6-二氟-吡啶-2-胺x-1c的合成
[0459]
向3,6-二氟吡啶-2-胺x-1b(0.60g,4.19mmol)在ch3cn(40ml)中的溶液中加入nbs(0.52g,2.93mmol),将该反应混合物在避光下在食物搅拌30min。加入nbs(0.52g,2.93mmol)的ch3cn(10ml)溶液,将该反应混合物在室温搅拌30min。通过tlc和lcms监测反应进程。完成后,该反应混合物用h2o(100ml)稀释,用etoac(160ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,5-10%在己烷中的etoac),得到5-溴-3,6-二氟吡啶-2-胺x-1c(0.70g),为灰白色固体。
[0460]
收率:79%
[0461]
碱性lc-ms方法2(es-):207(m-h)-,98%纯度。
[0462]1h nmr(400mhz,dmso-d6)δ6.86(brs,2h)7.82-7.91(m,1h)。
[0463]
步骤-4:5-溴-3,6-二氟-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-1d的合成
[0464]
在0℃向5-溴-3,6-二氟吡啶-2-胺x-1c(4.00g,19.0mmol)在dmac(40ml)中的溶液中分批加入nah(2.29g,57.1mmol),将该反应体系在相同温度搅拌30min。在0℃滴加对-甲氧基苄基氯(5.19ml,38.1mmol),将该反应混合物在室温搅拌2h。通过tlc监测反应进程。完成后,将该反应混合物冷却至0℃,用饱和nh4cl(20ml)淬灭,倾入h2o(60ml),用etoac(3
×
60ml)萃取。分离有机层,用盐水(2
×
80ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,4%在己烷中的etoac),得到5-溴-3,6-二氟-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-1d(8.2g,96%),为灰白色固体。
[0465]
收率:96%。
[0466]1h nmr(400mhz,dmso-d6)δ3.72(s,6h)4.57(s,4h)6.87-6.89(m,4h)7.16-7.18(m,4h)7.99-8.07(m,1h)。
[0467]
步骤-5:3,6-二氟-n,n-双[(4-甲氧基苯基)甲基]-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼环戊烷-2-基)吡啶-2-胺x-1e的合成:
[0468]
在室温向5-溴-3,6-二氟-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-1d(4.00g,8.90mmol)在二噁烷(160ml)中的溶液中加入双(频哪醇合)二硼(4.52g,17.8mmol)和koac(3.06g,31.2mmol),将该反应混合物用氩气吹扫20min,随后添加pdcl2(dppf)(0.65g,0.89mmol)。将该反应混合物用氩气吹扫10min,在100℃加热16h。通过tlc监测反应进程。完成后,冷却该反应混合物,通过硅藻土垫过滤,用etoac(2
×
80ml)洗涤。真空浓缩滤液,用h2o(100ml)稀释残余物,用etoac(2
×
80ml)萃取。分离有机层,用盐水(2
×
100ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,8%在己烷中的etoac),得到3,6-二氟-n,n-双[(4-甲氧基苯基)甲基]-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼环戊烷-2-基)吡啶-2-胺x-1e(2.60g),为灰白色固体。
[0469]
收率:59%。
[0470]1h nmr(400mhz,dmso-d6)δ1.26(s,12h)3.73(s,6h)4.64(s,4h)6.89(d,j=8.31hz,4h)7.17(d,j=8.80hz,4h)7.51-7.56(m,1h)。
[0471]
步骤-6:6-[双[(4-甲氧基苯基)甲基]氨基]-2,5-二氟-吡啶-3-醇x-1f的合成:
[0472]
在0℃向3,6-二氟-n,n-双[(4-甲氧基苯基)甲基]-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼环戊烷-2-基)吡啶-2-胺x-1e(2.50g,5.04mmol)在thf(30ml)中的溶液中加入30%h2o2的h2o溶液(10ml),将该反应混合物在相同温度搅拌15min。将该反应混合物在室温搅拌1.5h。通过tlc和lcms监测反应进程。在0℃将该反应混合物倾入5%na2s2o3的冷h2o溶液(250ml),用h2o(100ml)稀释,用etoac(2
×
100ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到6-[双[(4-甲氧基苯基)甲基]氨基]-2,5-二氟-吡啶-3-醇x-1f(1.74g粗制品),为黄色半固体。
[0473]
将该化合物原样用于下一步反应,未经进一步纯化。
[0474]
碱性lc-ms方法2(es
+
):387(m+h)
+
,93%纯度。
[0475]1h nmr(400mhz,dmso-d6)δ3.71(s,6h)4.32(s,4h)6.86(d,j=8.80hz,4h)7.14(d,j=8.31hz,4h)7.22-7.27(m,1h)9.84(s,1h)。
[0476]
步骤-7:3,6-二氟-5-(2-氟乙氧基)-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-1g的合成:
[0477]
在室温向6-[双[(4-甲氧基苯基)甲基]氨基]-2,5-二氟-吡啶-3-醇x-1f(0.70g,1.68mmol)在dmf(13ml)中的溶液中加入k2co3(0.70g,5.04mmol)和1-溴-2-氟乙烷(0.43g,3.36mmol)。将该反应混合物在微波中在80℃加热15min。通过tlc和lcms监测反应进程。完成后,将该反应混合物冷却至室温,倾入h2o(30ml),用etoac(3
×
25ml)萃取。分离有机层,用盐水(2
×
30ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,25%在己烷中的etoac),得到3,6-二氟-5-(2-氟乙氧基)-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-1g(0.52g),为棕色液体。
[0478]
收率:71%。
[0479]
碱性lc-ms方法2(es
+
):433(m+h)
+
,99%纯度。
[0480]1h nmr(400mhz,dmso-d6)δ3.72(s,6h)4.21-4.25(m,1h)4.29-4.32(m,1h)4.42(s,4h)4.63-4.66(m,1h)4.75-4.78(m,1h)6.87(d,j=8.86hz,4h)7.15(d,j=8.37hz,4h)7.69-7.76(m,1h)。
[0481]
步骤-5:3,6-二氟-5-(2-氟乙氧基)吡啶-2-胺x-1的合成:
[0482]
在0℃向3,6-二氟-5-(2-氟乙氧基)-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-1g(0.50g,1.15mmol)中加入tfa(5ml),将该反应混合物在相同温度搅拌30min。将该反应混合物在室温搅拌2h。通过tlc和lcms监测反应进程。完成后,真空浓缩该反应混合物。残余物用h2o(25ml)稀释,用nahco3水溶液(10ml)碱化,用etoac(2
×
25ml)萃取。分离有机层,用盐水(2
×
40ml)洗涤,用无水na2so4干燥,真空浓缩。通过与et2o(10ml)一起研磨纯化得到的粗制品,真空干燥,得到3,6-二氟-5-(2-氟乙氧基)吡啶-2-胺x-1(0.258g),为灰白色固体。
[0483]
收率:80%。
[0484]
碱性lc-ms方法2(es
+
):193(m+h)
+
,69%纯度。
[0485]1h nmr(400mhz,dmso-d6)δ4.13-4.17(m,1h)4.20-4.25(m,1h)4.59-4.64(m,1h)
4.71-4.76(m,1h)6.10(s,2h)7.57-7.62(m,1h)。
[0486]
a.2. 3-氟-5-(2-氟乙氧基)吡啶-2-胺x-2的合成:
[0487][0488]
步骤-1:5-溴-3-氟-n,n-双(4-甲氧基苄基)吡啶-2-胺x-2a的合成:
[0489]
在0℃向5-溴-3-氟-吡啶-2-胺(2.00g,10.5mmol)在dmac(30ml)中的溶液中分批加入nah(1.26g,31.4mmol),将该反应混合物在相同温度搅拌30min。在0℃加入对-甲氧基苄基氯(2.85ml,20.9mmol),将该反应混合物在室温搅拌2h。通过tlc和lcms监测反应进程。完成后,在0℃冷却该反应混合物,用饱和nh4cl溶液(200ml)淬灭,倾入冰h2o(400ml),用etoac(2
×
300ml)萃取。分离有机层,用盐水(2
×
100ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,3%在己烷中的etoac),得到5-溴-3-氟-n,n-双(4-甲氧基苄基)吡啶-2-胺x-2a(4.10g),为淡黄色半固体。
[0490]
收率:87%
[0491]
碱性lc-ms方法2(es
+
):431(m+h)
+
,95%纯度。
[0492]1h nmr(400mhz,dmso-d6)δ3.72(s,6h),4.57(s,4h),6.87(d,j=8.31hz,4h),7.15(d,j=8.80hz,4h),7.81(dd,j=12.96,1.71hz,1h),8.06(s,1h)。
[0493]
步骤-2:3-氟-n,n-双(4-甲氧基苄基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼环戊烷-2-基)吡啶-2-胺x-2b的合成:
[0494]
在室温向5-溴-3-氟-n,n-双(4-甲氧基苄基)吡啶-2-胺x-2a(2.00g,4.43mmol)在二噁烷(60ml)中的溶液中加入双(频哪醇合)二硼(2.25g,8.86mmol)和koac(1.52g,15.5mmol),将该反应混合物用氩气吹扫20min,随后添加pdcl2(dppf)(0.32g,0.44mmol)。
将该反应混合物用氩气吹扫10min,在100℃加热16h。通过tlc监测反应进程。完成后,将该反应混合物冷却至室温,通过硅藻土垫过滤,用etoac(2
×
300ml)洗涤,真空浓缩滤液。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,5-10%在己烷中的etoac)。将得到的产物在戊烷(10ml)中搅拌,滗析,真空干燥,得到3-氟-n,n-双(4-甲氧基苄基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼环戊烷-2-基)吡啶-2-胺x-2b(1.31g),为灰白色固体。
[0495]
收率:56%
[0496]
碱性lc-ms方法2(es
+
):479(m+h)
+
,90%纯度。
[0497]1h nmr(400mhz,dmso-d6)δ1.27(s,12h),3.72(s,6h),4.65(s,4h),6.87(d,j=8.80hz,4h),7.15(d,j=8.31hz,4h),7.44(d,j=15.16hz,1h),8.15(s,1h)。
[0498]
步骤-3:6-(双(4-甲氧基苄基)氨基)-5-氟吡啶-3-醇x-2c的合成:
[0499]
在0℃向x-2b(1.31g,2.46mmol)在thf(20ml)中的溶液中加入30%h2o2的h2o溶液(7ml),将该反应混合物在相同温度搅拌15min。将该反应混合物在室温搅拌2h。通过tlc和lcms监测反应进程。在0℃将该反应混合物倾入冷5%na2s2o3的h2o溶液(350ml),用h2o(100ml)稀释,用et2o(2
×
200ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩(低温),得到6-(双(4-甲氧基苄基)氨基)-5-氟吡啶-3-醇x-2c(0.90g粗制品),为淡黄色半固体。
[0500]
将该化合物原样用于下一步反应,未经进一步纯化。
[0501]
碱性lc-ms方法2(es
+
):369(m+h)
+
,94%纯度。
[0502]
步骤-4:3-氟-5-(2-氟乙氧基)-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-2d的合成:
[0503]
在室温向6-(双(4-甲氧基苄基)氨基)-5-氟吡啶-3-醇x-2c(1.70g,4.43mmol)在dmf(10ml)中的溶液中加入k2co3(0.77g,5.56mmol)和1-溴-2-氟乙烷(0.67g,5.31mmol)。将该反应混合物在微波中在70℃加热过夜。通过tlc和lcms监测反应进程。完成后,将该反应混合物冷却至室温,倾入h2o(100ml),用etoac(2
×
50ml)萃取。分离有机层,用冰冷水(2
×
25ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,5-8%在己烷中的etoac),得到3-氟-5-(2-氟乙氧基)-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-2d(0.80g),为棕色液体。
[0504]
收率:43%
[0505]
碱性lc-ms方法2(es
+
):415(m+h)
+
,99%纯度。
[0506]1h nmr(400mhz,dmso-d6)δ3.71(s,6h),4.14-4.22(m,1h),4.25-4.29(m,1h),4.40(s,4h),4.63-4.68(m,1h),4.74-4.80(m,1h),6.85(d,j=8.80hz,4h),7.14(d,j=8.31hz,4h),7.39(dd,j=14.18,2.45hz,1h),7.78(d,j=1.96hz,1h)。
[0507]
步骤-5:3-氟-5-(2-氟乙氧基)吡啶-2-胺x-2的合成:
[0508]
向x-2d(800mg,1.92mmol)在meoh(3ml)中的溶液中加入氢氧化钯(160mg),将该反应混合物在室温在氢气压力下搅拌6h。通过tlc和lcms监测反应进程。完成后,通过硅藻土过滤该反应混合物,用meoh(2
×
25ml)洗涤,真空浓缩滤液。通过柱色谱法纯化残余物(硅胶,100-200目,10至15%在己烷中的etoac),得到3-氟-5-(2-氟乙氧基)吡啶-2-胺x-2(100mg),为棕色固体。
[0509]
收率:29%
[0510]
碱性lc-ms方法2(es
+
):175(m+h)
+
,97%纯度。
[0511]1h nmr(400mhz,dmso-d6)δ4.11-4.16(m,1h),4.19-4.23(m,1h),4.60-4.64(m,1h),4.72-4.77(m,1h),5.72(s,2h),7.27(dd,j=12.23,2.45hz,1h),7.59(d,j=2.45hz,1h)。
[0512]
a.3. 3-氟-5-(2,2-二氟乙氧基)吡啶-2-胺x-3的合成:
[0513][0514]
步骤-1:5-(2,2-二氟乙氧基)-3-氟-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-3a的合成:
[0515]
在室温向6-(双(4-甲氧基苄基)氨基)-5-氟吡啶-3-醇x-2c(1.40g,3.69mmol)和2,2-二氟乙醇(0.66g,8.11mmol)在甲苯(25ml)中的溶液中缓慢地加入(氰基亚甲基)三丁基膦(1.96g,8.11mmol)。将该反应混合物在100℃加热4h。通过tlc监测反应进程。完成后,将该反应混合物冷却至室温,倾入h2o(80ml),用etoac(3
×
100ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,10%在己烷中的etoac),得到5-(2,2-二氟乙氧基)-3-氟-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-3a(1.22g),为淡黄色液体。
[0516]
收率:59%
[0517]
碱性lc-ms方法2(es
+
):433(m+h)
+
,77%纯度。
[0518]
步骤-2:3-氟-5-(2,2-二氟乙氧基)吡啶-2-胺x-3的合成:
[0519]
向x-3a(1.20g,2.19mmol)在meoh(30ml)中的溶液中加入碳负载氢氧化钯(500mg),将该反应混合物在室温在氢气压力下搅拌6h。通过tlc和lcms监测反应进程。完成后,通过硅藻土过滤该反应混合物,用meoh(2
×
50ml)洗涤,真空浓缩滤液,得到3-氟-5-(2,2-二氟乙氧基)吡啶-2-胺x-3(590mg),为淡黄色半固体。
[0520]
将该化合物原样用于下一步反应,未经进一步纯化。
[0521]
收率:77%
[0522]
碱性lc-ms方法2(es
+
):193(m+h)
+
,55%纯度。
[0523]
a.4. 5-(2,2-二氟乙氧基)-3,6-二氟吡啶-2-胺x-4的合成:
[0524]
[0525]
步骤-1:5-(2,2-二氟乙氧基)-3,6-二氟-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-4a的合成
[0526]
在室温向6-[双[(4-甲氧基苯基)甲基]氨基]-2,5-二氟-吡啶-3-醇x-1f(100mg,0.16mmol)、2,2-二氟乙醇(27mg,0.33mmol)和三苯膦(218mg,0.83mmol)在thf(5ml)中的溶液中缓慢地加入偶氮二羧酸二异丙酯(168mg,0.829mmol)。将该反应混合物在室温搅拌1h。通过tlc监测反应进程。完成后,用h2o(20ml)稀释该反应混合物,用etoac(20ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到5-(2,2-二氟乙氧基)-3,6-二氟-n,n-双[(4-甲氧基苯基)甲基]吡啶-2-胺x-4a(20mg),为半固体。
[0527]
将该化合物原样用于下一步反应,未经进一步纯化。
[0528]
收率:27%
[0529]1h nmr(400mhz,dmso-d6)δ3.72(s,6h),4.44(s,4h),4.79-4.85(m,2h),6.01-6.31(m,1h),6.87(d,j=8.31hz,4h),7.15(d,j=8.31hz,4h),7.76-7.81(m,1h)。
[0530]
步骤-2:5-(2,2-二氟乙氧基)-3,6-二氟吡啶-2-胺x-4的合成:
[0531]
向x-4a(540mg,1.20mmol)在meoh(10ml)中的溶液中加入碳负载氢氧化钯(54mg),将该反应混合物在室温在氢气压力下搅拌1h。通过tlc和lcms监测反应进程。完成后,通过硅藻土过滤该反应混合物,用meoh(2
×
50ml)洗涤,真空浓缩滤液,得到5-(2,2-二氟乙氧基)-3,6-二氟吡啶-2-胺x-4(300mg),为棕色固体。
[0532]
将该化合物原样用于下一步反应,未经进一步纯化。
[0533]
收率:71%
[0534]
碱性lc-ms方法2(es
+
):211(m+h)
+
,60%纯度。
[0535]
a.5. 5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-胺x-5的合成:
[0536][0537]
向5-(2,2-二氟乙氧基)-3,6-二氟吡啶-2-胺x-4(0.39g,1.74mmol)在meoh(14ml)中的溶液中加入naome(0.48g,8.79mmol),将该反应混合物在100℃加热30h。通过tlc和lcms监测反应进程。完成后,真空浓缩该反应混合物。用h2o(500ml)稀释残余物,用etoac(600ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-胺x-5(0.37g),为棕色固体。
[0538]
将该化合物原样用于下一步反应,未经进一步纯化。
[0539]
收率:67%
[0540]
碱性lc-ms方法2(es-):221(m-h)-,70%纯度。
[0541]
1h nmr(400mhz,dmso-d6)δ3.78(s,3h)4.13(td,j=14.67,3.42hz,2h)5.72(brs,2h)6.13-6.45(m,1h)7.34(d,j=10.76hz,1h)。
[0542]
a.6. 5-(二氟甲氧基)-3-甲氧基吡啶-2-胺x-6的合成:
[0543][0544]
步骤-1:5-溴-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-6a的合成:
[0545]
在0℃向5-溴-3-甲氧基-吡啶-2-胺(5.00g,24.6mmol)在dmf(75ml)中的溶液中分批加入nah(2.95g,73.9mmol),将该反应混合物在相同温度搅拌30min。在0℃滴加对-甲氧基苄基氯(6.71ml,49.3mmol),将该反应混合物在室温搅拌2h。通过tlc和lcms监测反应进程。完成后,在0℃冷却该反应混合物,用冷h2o(250ml)和饱和nh4cl溶液(250ml)淬灭,用etoac(2
×
500ml)萃取。分离有机层,用盐水(2
×
100ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,25%在己烷中的etoac),得到5-溴-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-6a(9.65g),为灰白色固体。
[0546]
收率:75%
[0547]
碱性lc-ms方法2(es
+
):443(m+h)
+
,85%纯度。
[0548]1h nmr(400mhz,dmso-d6)δ3.69(s,6h)3.82(s,3h)4.42(s,4h)6.79-6.85(m,4h)7.11(d,j=8.31hz,4h)7.39(d,j=1.96hz,1h)7.76(d,j=1.96hz,1h)。
[0549]
步骤-2:3-甲氧基-n,n-双(4-甲氧基苄基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼环戊烷-2-基)吡啶-2-胺x-6b的合成:
[0550]
在室温向5-溴-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-6a(9.60g,18.3mmol)在二噁烷(300ml)中的溶液中加入双(频哪醇合)二硼(9.31g,36.7mmol)和koac(6.30g,64.1mmol),将该反应混合物用氩气吹扫20min,随后添加pdcl2(dppf)(1.34g,1.83mmol)。将该反应混合物用氩气吹扫10min,在100℃加热16h。通过tlc和lcms监测反应进程。完成后,将该反应混合物冷却至室温,通过硅藻土垫过滤,用etoac(2
×
300ml)洗涤,
真空浓缩滤液。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,5-10%在己烷中的etoac)。将得到的产物与戊烷(50ml一起搅拌,滗析,真空干燥,得到3-甲氧基-n,n-双(4-甲氧基苄基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼环戊烷-2-基)吡啶-2-胺x-6b(5.10g),为灰白色固体。
[0551]
收率:47%
[0552]
碱性lc-ms方法2(es
+
):491(m+h)
+
,83%纯度。
[0553]1h nmr(400mhz,dmso-d6)δ1.26(s,12h)3.69(s,6h)3.77(s,3h)4.55(s,4h)6.82(d,j=8.80hz,4h)7.11(d,j=8.31hz,4h)7.21(s,1h)7.94(s,1h)。
[0554]
步骤-3:6-(双(4-甲氧基苄基)氨基)-5-甲氧基吡啶-3-醇x-6c的合成:
[0555]
在0℃向3-甲氧基-n,n-双(4-甲氧基苄基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂硼环戊烷-2-基)吡啶-2-胺x-6b(5.00g,8.49mmol)在thf(100ml)中的溶液中缓慢地加入30%h2o2的h2o溶液(40ml)。15min后,将该反应混合物在室温搅拌2h。通过tlc和lcms监测反应进程。完成后,在0℃将该反应混合物倾入冷6%na2s2o3的h2o溶液(600ml),用h2o(500ml)稀释,用et2o(2
×
400ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩(在低温,20-25℃)。用戊烷(15ml)洗涤残余物,得到6-(双(4-甲氧基苄基)氨基)-5-甲氧基吡啶-3-醇x-6c(3.50g粗制品),为淡黄色固体。
[0556]
将该化合物原样用于下一步反应,未经进一步纯化。
[0557]
碱性lc-ms方法2(es
+
):381(m+h)
+
,84%纯度。
[0558]1h nmr(400mhz,dmso-d6)δ3.69(s,6h)3.81(s,3h)4.18(s,4h)6.77(d,j=2.45hz,1h)6.80(d,j=8.31hz,4h)7.12(d,j=8.80hz,4h)7.28(d,j=2.45hz,1h)9.21(s,1h)。
[0559]
步骤-4:5-(二氟甲氧基)-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-6d的合成:
[0560]
在0℃向6-(双(4-甲氧基苄基)氨基)-5-甲氧基吡啶-3-醇x-6c(3.30g,7.31mmol)在ch3cn(60ml)中的溶液中滴加koh(2.26g,40.2mmol)的h2o溶液(20ml),随后在0℃添加二乙基膦酸溴二氟甲酯(10.7g,40.2mmol)。15min后,将该反应混合物在室温搅拌2h。通过tlc和lcms监测反应进程。完成后,用h2o(200ml)使该反应混合物淬灭,用etoac(2
×
250ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,5%在己烷中的etoac),得到5-(二氟甲氧基)-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-6d(3.21g),为淡黄色半固体。
[0561]
收率:92%
[0562]
碱性lc-ms方法2(es
+
):431(m+h)
+
,90%纯度。
[0563]1h nmr(400mhz,dmso-d6)δ3.70(s,6h)3.85(s,3h)4.39(s,4h)6.83(d,j=8.80hz,4h)7.11-7.16(m,4h)7.14(t,j=74hz,1h)7.19(d,j=2.45hz,1h)7.62(d,j=1.96hz,1h)。
[0564]
步骤-5:5-(二氟甲氧基)-3-甲氧基吡啶-2-胺x-6的合成:
[0565]
在0℃向5-(二氟甲氧基)-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-6d(2.50g,5.26mmol)中加入tfa(15ml),将该反应混合物在室温搅拌4h。通过tlc和lcms监测反应进程。完成后,真空浓缩该反应混合物。用h2o(400ml)和nahco3饱和溶液(250ml)稀释残余物,用etoac(2
×
300ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过急骤色谱法纯化得到的粗制品(40%在己烷中的etoac),得到5-(二氟甲氧基)-3-甲氧基吡啶-2-胺x-6
(0.52g),为灰白色固体。
[0566]
收率:52%
[0567]
碱性lc-ms方法2(es
+
):191(m+h)
+
,99%纯度。
[0568]1h nmr(400mhz,dmso-d6)δ3.79(s,3h)5.77(s,2h)6.98(d,j=1.96hz,1h)7.01(t,j=74hz,1h)7.42(d,j=1.96hz,1h)。
[0569]
a.7. 5-(2,2-二氟乙氧基)-3-甲氧基吡啶-2-胺x-7的合成:
[0570][0571]
步骤-1:5-(2,2-二氟乙氧基)-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-7a的合成:
[0572]
在室温向6-(双(4-甲氧基苄基)氨基)-5-甲氧基吡啶-3-醇x-6c(2.80g,7.01mmol)和2,2-二氟乙醇(0.89ml,14.0mmol)在甲苯(30ml)中的溶液中缓慢地加入氰基亚甲基三丁基膦(3.67ml,14.0mmol),将该反应混合物在100℃加热2h。通过tlc和lcms监测反应进程。完成后,用h2o(30ml)稀释该反应混合物,用etoac(3
×
40ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,30%在己烷中的etoac),得到5-(2,2-二氟乙氧基)-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-7a(2.10g),为淡黄色油状物。
[0573]
收率:67%
[0574]
碱性lc-ms方法2(es
+
):445(m+h)
+
,99%纯度。
[0575]1h nmr(400mhz,dmso-d6)δ3.68(s,6h)3.85(s,3h)4.24-4.34(m,6h)6.19-6.49(m,1h)6.80(d,j=8.80hz,4h)7.03(d,j=1.96hz,1h)7.11(d,j=8.31hz,4h)7.48(d,j=2.45hz,1h)。
[0576]
步骤-2:5-(2,2-二氟乙氧基)-3-甲氧基吡啶-2-胺x-7的合成:
[0577]
在0℃向dcm(15ml)中的5-(2,2-二氟乙氧基)-3-甲氧基-n,n-双(4-甲氧基苄基)吡啶-2-胺x-7a(1.70g,3.82mmol)中滴加tfa(10ml),将该反应混合物在室温搅拌4h。通过tlc和lcms监测反应进程。完成后,真空浓缩该反应混合物。用h2o(20ml)、饱和nahco3(20ml)稀释残余物,用etoac(2
×
30ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过急骤色谱法纯化得到的粗制品(30%在己烷中的etoac),得到5-(2,2-二氟乙氧基)-3-甲氧基吡啶-2-胺x-7(0.605g),为淡黄色固体。
[0578]
收率:74%
[0579]
碱性lc-ms方法2(es
+
):205(m+h)
+
,99%纯度。
[0580]1h nmr(400mhz,dmso-d6)δ3.77(s,3h)4.23(td,j=14.80,3.67hz,2h)5.33(s,2h)6.18-6.50(m,1h)6.88(d,j=2.45hz,1h)7.31(d,j=2.45hz,1h)。
[0581]
a.8. 5-(2-(二氟甲氧基)乙氧基)-3-氟-6-甲氧基吡啶-2-胺x-8的合成:
[0582][0583]
步骤-1:5-(2-(二氟甲氧基)乙氧基)-3,6-二氟-n,n-双(4-甲氧基苄基)吡啶-2-胺x-8a的合成:
[0584]
在室温向6-[双[(4-甲氧基苯基)甲基]氨基]-2,5-二氟-吡啶-3-醇x-1f(0.80g,1.52mmol)在dmf(10ml)中的溶液中加入k2co3(0.63g,4.56mmol)和1-溴-2-(二氟甲氧基)乙烷(0.29g,1.67mmol)。将该反应混合物在微波中在85℃加热1.2h。通过tlc和lcms监测反应进程。完成后,将该反应混合物冷却至室温,倾入h2o(60ml),用etoac(3
×
40ml)萃取。分离有机层,用盐水(2
×
50ml)洗涤,用无水na2so4干燥,真空浓缩。用0.70g和2
×
0.80g重复该反应,将从4次反应得到的粗制品在dcm(200ml)中击打,通过柱色谱法纯化(硅胶,100-200目,8%在己烷中的etoac),得到5-(2-(二氟甲氧基)乙氧基)-3,6-二氟-n,n-双(4-甲氧基苄基)吡啶-2-胺x-8a(1.31g),为棕色液体。
[0585]
收率:34%
[0586]
碱性lc-ms方法2(es
+
):481(m+h)
+
,99%纯度。
[0587]
步骤-2:5-(2-(二氟甲氧基)乙氧基)-3,6-二氟吡啶-2-胺x-8b的合成:
[0588]
在室温向5-(2-(二氟甲氧基)乙氧基)-3,6-二氟-n,n-双(4-甲氧基苄基)吡啶-2-胺x-8a(1.30g,2.36mmol)在meoh(25ml)中的溶液中加入20%pd/c(0.13g),将该反应混合物在室温在氢气压力下搅拌2h。通过tlc和lcms监测反应进程。完成后,通过硅藻土垫过滤该反应混合物,用meoh(2
×
30ml)洗涤。真空浓缩滤液。通过用戊烷(2
×
20ml)洗涤纯化得到的粗制品,得到5-(2-(二氟甲氧基)乙氧基)-3,6-二氟吡啶-2-胺x-8b(0.525g),为浅棕色固体。
[0589]
收率:92%
[0590]
碱性lc-ms方法2(es
+
):241(m+h)
+
,99%纯度。
[0591]1h nmr(400mhz,dmso-d6)δ4.03-4.07(m,2h)4.08-4.10(m,2h)6.07(s,2h)6.68(t,j=76hz 1h)7.53-7.57(m,1h)。
[0592]
步骤-3:5-(2-(二氟甲氧基)乙氧基)-3-氟-6-甲氧基吡啶-2-胺x-8的合成
[0593]
在0℃向5-(2-(二氟甲氧基)乙氧基)-3,6-二氟吡啶-2-胺x-8b(0.70g,2.91mmol)
在meoh(15ml)中的溶液中加入naome(25%的meoh溶液,1.57ml,7.29mmol),将该反应混合物在70℃加热24h。通过tlc和lcms监测反应进程。完成后,真空浓缩该反应混合物。用h2o(20ml)稀释残余物,用etoac(3
×
25ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过combi-急骤色谱法纯化得到的粗制品(10%在己烷中的etoac),得到5-(2-(二氟甲氧基)乙氧基)-3-氟-6-甲氧基吡啶-2-胺x-8(0.45g),为淡黄色固体。
[0594]
收率:59%
[0595]
碱性lc-ms方法2(es
+
):253(m+h)
+
,94%纯度。
[0596]1h nmr(400mhz,dmso-d6)δ3.77(s,3h)4.03-4.06(m,4h)5.61(s,2h)6.72(t,j=76hz,1h)7.26(d,j=11.25hz,1h)。
[0597]
b.式xi的中间体的合成
[0598]
b.1. 6,7-二氯-1h-吲哚xi-1的合成:
[0599][0600]
在-78℃向1,2-二氯-3-硝基-苯(4.00g,20.8mmol)在thf(60ml)中的溶液中加入溴化乙烯基镁(1m,83.3ml,83.3mmol),将该反应混合物在相同温度搅拌3h。通过tlc和lcms监测反应进程。完成后,用饱和nh4cl(80ml)使该反应混合物淬灭,用etoac(3
×
100ml)萃取。分离有机层,用盐水洗涤(100ml),用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,2%在己烷中的etoac),得到6,7-二氯-1h-吲哚xi-1(1.50g),为灰白色固体。
[0601]
收率:37%。
[0602]
碱性lcms方法2(es
+
):186(m+h)
+
,95%纯度。
[0603]1h nmr(400mhz,dmso-d6)δ6.56-6.57(m,1h),7.30(d,j=8.4hz,1h),7.43(t,j=2.4hz,1h),7.48(d,j=8.4hz,1h),11.62(brs,1h)。
[0604]
b.2. 6-氯-7-环丙氧基-1h-吲哚xi-2的合成:
[0605][0606]
步骤-1:1-氯-2-环丙氧基-3-硝基苯xi-2a的合成:
[0607]
在惰性气体气氛中在0℃向搅拌的nah(0.91g,22.8mmol)在thf(30ml)中的混悬液中加入环丙醇(1.08ml,17.1mmol),将该反应混合物在相同温度搅拌30min。在0℃滴加1-氯-2-氟-3-硝基-苯(2.00g,11.4mmol)在thf(10ml)中的溶液,将该反应混合物在75℃加热3h。通过tlc监测反应进程。完成后,在室温冷却该反应混合物,倾入碎冰,用etoac(2
×
7.45(m,1h)7.57(d,j=8.31hz,1h)11.48(brs,1h)
[0622]
步骤-2:7-溴-6-氯-1-(苯磺酰基)-1h-吲哚xi-3b的合成:
[0623]
在0℃向搅拌的nah(2.42g,60.5mmol)在干thf(30ml)中的混悬液中分批加入7-溴-6-氯-1h-吲哚xi-3a(7.00g,30.2mmol),将该反应混合物搅拌20min。滴加苯磺酰氯(5.79ml,45.4mmol)在thf(10ml)中的溶液,将该反应混合物在室温搅拌16h。通过tlc和lcms监测反应进程。完成后,用etoac(30ml)稀释该反应混合物,用饱和nahco3溶液(25ml)洗涤。分离有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的有机粗制品(硅胶,100-200目,20%在己烷中的etoac),得到7-溴-6-氯-1-(苯磺酰基)-1h-吲哚xi-3b(7.50g),为灰白色固体。
[0624]
收率:54%
[0625]
碱性lcms方法2(es
+
):370(m+h)
+
,81%纯度。
[0626]1h nmr(400mhz,dmso-d6)δ6.88(d,j=3.42hz,1h),7.44(d,j=8.80hz,1h),7.58-7.64(m,3h),7.70-7.75(m,1h),7.87(d,j=3.42hz,1h),8.01(d,j=7.82hz,2h)。
[0627]
步骤-3:6-氯-1-(苯磺酰基)-7-乙烯基-1h-吲哚xi-3c的合成:
[0628]
向7-溴-6-氯-1-(苯磺酰基)-1h-吲哚xi-3b(4.00g,8.72mmol)在dmf(15ml)中的溶液中加入三丁基(乙烯基)锡烷(3.07ml,10.5mmol)、三苯膦(0.11g,0.44mmol)和licl(1.11g,26.2mmol),将该反应混合物用氩气吹扫10min。加入pdcl2(pph3)2(0.31g,0.44mmol),将该反应混合物在密封试管中在100℃加热16h。通过tlc监测反应进程。完成后,通过硅藻土垫过滤该反应混合物,用etoac(2
×
40ml)洗涤。用盐水(20ml)洗涤滤液,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,20%在己烷中的etoac),得到6-氯-1-(苯磺酰基)-7-乙烯基-1h-吲哚xi-3c(2.80g),为淡黄色固体。
[0629]
收率:98%
[0630]1h nmr(400mhz,dmso-d6)δ4.88(d,j=18.10hz,1h),5.29(d,j=12.23hz,1h),6.81-6.89(m,1h),6.93(d,j=3.91hz,1h),7.39(d,j=8.31hz,1h),7.54-7.63(m,3h),7.70(d,j=7.34hz,1h),7.74(d,j=7.83hz,2h),7.93(d,j=3.42hz,1h)。
[0631]
步骤-4:6-氯-1-(苯磺酰基)-1h-吲哚-7-甲醛xi-3d的合成:
[0632]
在0℃向6-氯-1-(苯磺酰基)-7-乙烯基-1h-吲哚xi-3c(2.50g,7.63mmol)在thf(20ml)和h2o(5ml)中的溶液中加入oso4(0.04m的水溶液,3.77ml,0.15mmol),将该反应混合物搅拌30min。在0℃加入naio4(4.08g,19.1mmol),将该反应混合物在室温搅拌4h。通过tlc监测反应进程。完成后,通过硅藻土垫过滤该反应混合物,用etoac(2
×
20ml)洗涤。用盐水(20ml)洗涤滤液,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,20%在己烷中的etoac),得到6-氯-1-(苯磺酰基)-1h-吲哚-7-甲醛xi-3d(0.60g),为淡黄色固体。
[0633]
收率:24%
[0634]1h nmr(400mhz,dmso-d6)δ7.01(d,j=3.91hz,1h),7.48(d,j=8.31hz,1h),7.56-7.62(m,2h),7.68-7.75(m,3h),7.78(d,j=8.80hz,1h),7.93(d,j=3.91hz,1h),10.43(s,1h)。
[0635]
步骤-5:6-氯-7-(二氟甲基)-1-(苯磺酰基)-1h-吲哚xi-3的合成:
[0636]
在0℃向6-氯-1-(苯磺酰基)-1h-吲哚-7-甲醛xi-3d(0.60g,1.84mmol)在dcm
(15ml)中的溶液中加入氟化二乙基氨基硫(1.06ml,7.36mmol),将该反应混合物在室温搅拌16h。通过tlc监测反应进程。完成后,将该反应混合物倾入冰h2o(15ml),用饱和nahco3溶液(15ml)碱化,etoac(2
×
20ml)用萃取。分离有机层,用盐水(15ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,20%在己烷中的etoac),得到6-氯-7-(二氟甲基)-1-(苯磺酰基)-1h-吲哚xi-3(0.55g),为灰白色固体。
[0637]
收率:81%
[0638]1h nmr(400mhz,cdcl3)δ6.67(d,j=3.91hz,1h),7.35-7.44(m,4h),7.50-7.55(m,1h),7.58(d,j=8.31hz,2h),7.62(d,j=3.42hz,1h),7.86(t,j=52hz,1h)。
[0639]
b.4. 6-氯-7-氟-1h-吲哚xi-4的合成:
[0640][0641]
在-78℃向1-氯-2-氟-3-硝基-苯(2.50g,14.2mmol)在thf(50ml)中的溶液中加入溴化乙烯基镁(5.61g,42.7mmol),将该反应混合物在相同温度搅拌1h。通过tlc和lcms监测反应进程。完成后,用饱和nh4cl(100ml)使该反应混合物淬灭,用h2o(400ml)稀释,用etoac(500ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,5%在己烷中的etoac),得到6-氯-7-氟-1h-吲哚xi-4(0.60g),为红色液体。
[0642]
收率:17%
[0643]
碱性lcms方法2(es-):168(m-h)-,66%纯度。
[0644]
b.5. 6-(二氟甲基)-7-氟-1h-吲哚xi-5的合成:
[0645][0646]
步骤-1:1-(二氟甲基)-2-氟-3-硝基-苯xi-5a的合成:
[0647]
在0℃向2-氟-3-硝基-苯甲醛(2.0g,11.8mmol)在dcm(20ml)中的溶液中加入氟化二乙基氨基硫(7.63g,47.3mmol),将该反应混合物在室温搅拌4h。通过tlc监测反应进程。完成后,将该反应混合物倾入冰冷饱和nahco3溶液(50ml),用dcm(2
×
200ml)萃取。分离有机层,用盐水(15ml)洗涤,用无水na2so4干燥,真空浓缩,得到1-(二氟甲基)-2-氟-3-硝基-苯xi-5a(2.23g),为浅棕色液体。
[0648]
将该化合物原样用于下一步反应,未经进一步纯化。
[0649]
收率:96%
[0650]1h nmr(400mhz,dmso-d6)δ7.35(t,j=52hz,1h),7.60(t,j=8.07hz,1h),8.05(t,j=6.85hz,1h),8.35(t,j=7.82hz,1h)。
[0651]
步骤-2:6-(二氟甲基)-7-氟-1h-吲哚xi-5的合成:
[0652]
在0℃向1-(二氟甲基)-2-氟-3-硝基-苯xi-5a(2.21g,11.2mmol)在thf(20ml)中的溶液中加入溴化乙烯基镁(1m的thf溶液,45ml,45mmol),将该反应混合物在相同温度搅拌3h。通过tlc和lcms监测反应进程。完成后,在0℃用饱和nh4cl(40ml)使该反应混合物淬灭,用etoac(2
×
200ml)萃取。用盐水(30ml)洗涤有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,10%在己烷中的etoac),得到6-(二氟甲基)-7-氟-1h-吲哚xi-5(0.54g),为棕色油状物。
[0653]
收率:25%
[0654]
碱性lcms方法2(es-):184(m-h)-,95%纯度。
[0655]1h nmr(400mhz,dmso-d6)δ6.57-6.62(m,1h),7.14-7.17(m,1h),7.29(t,j=56hz 1h),7.48(d,j=8.19hz,1h),7.56(t,j=2.69hz,1h),11.94(br s,1h)。
[0656]
b.6. 1-(苯磺酰基)-7-氯-6-(二氟甲基)吲哚xi-6的合成:
[0657][0658]
步骤-1:6-溴-7-氯-1h-吲哚xi-6a的合成
[0659]
在-78℃向1-溴-2-氯-3-硝基-苯(5.0g,21.1mmol)在thf(80ml)中的溶液中滴加溴化乙烯基镁(1m的thf溶液,84.6ml,84.6mmol),将该反应混合物在相同温度搅拌3h。通过tlc和lcms监测反应进程。完成后,用饱和nh4cl(30ml)使该反应混合物淬灭,用etoac(3
×
100ml)萃取。分离有机层,用盐水(70ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,3to 5%在己烷中的etoac),得到6-溴-7-氯-1h-吲哚xi-6a(2.5g),为黄色固体。
[0660]
收率:49%
[0661]
碱性lcms方法2(es-):228(m-h)-,96%纯度。
[0662]
步骤-2:6-溴-7-氯-1-(苯磺酰基)-1h-吲哚xi-6b的合成:
[0663]
在0℃向搅拌的nah(0.87g,21.7mmol)在干thf(30ml)中的混悬液中分批加入6-溴-7-氯-1h-吲哚xi-6a(2.5g,10.8mmol),将该反应混合物搅拌10min。在0℃滴加苯磺酰氯(2.3g,13mmol),将该反应混合物在室温搅拌2h。通过tlc和lcms监测反应进程。完成后,将该反应混合物倾入冰冷水(100ml),用etoac(3
×
50ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到6-溴-7-氯-1-(苯磺酰基)-1h-吲哚xi-6b(3.0g),为浅棕色固体。
[0664]
将该化合物原样用于下一步反应,未经进一步纯化。
[0665]
收率:71%
[0666]
碱性lcms方法2(es-):368(m-h)-,95%纯度。
[0667]
步骤-3:7-氯-1-(苯磺酰基)-6-乙烯基-1h-吲哚xi-6c的合成:
[0668]
向6-溴-7-氯-1-(苯磺酰基)-1h-吲哚xi-6b(1.5g,3.86mmol)在dmf(10ml)中的溶液中加入三丁基(乙烯基)锡烷(1.47g,4.63mmol)、三苯膦(50mg,0.19mmol)和licl(0.49g,11.6mmol),将该反应混合物用氩气吹扫10min。加入pdcl2(pph3)2(0.13g,0.19mmol),将该反应混合物在密封试管中在100℃加热16h。通过tlc监测反应进程。完成后,通过硅藻土垫过滤该反应混合物,用etoac(2
×
100ml)洗涤。用盐水(100ml)洗涤滤液,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,20%在己烷中的etoac),得到7-氯-1-(苯磺酰基)-6-乙烯基-1h-吲哚xi-6c(0.80g),为灰白色固体。
[0669]
收率:64%
[0670]
碱性lcms方法2(es
+
):318(m+h)
+
,98%纯度。
[0671]
步骤-4:1-(苯磺酰基)-7-氯-吲哚-6-甲醛xi-6d的合成:
[0672]
在0℃向7-氯-1-(苯磺酰基)-6-乙烯基-1h-吲哚xi-6c(1.40g,4.19mmol)在thf(70ml)和h2o(15ml)中的溶液中加入oso4(0.04m的水溶液,2.07ml,0.08mmol),将该反应混合物搅拌3小时。在0℃加入naio4(2.69g,12.6mmol),将该反应混合物在室温搅拌8h。通过tlc监测反应进程。完成后,水(100ml)用稀释该反应混合物。过滤沉淀,真空干燥,得到1-(苯磺酰基)-7-氯-吲哚-6-甲醛xi-6d(1.30g),为棕色固体。
[0673]
将该化合物原样用于下一步反应,未经进一步纯化。
[0674]
收率:82%
[0675]
碱性lcms方法2(es
+
):320(m+h)
+
,82%纯度。
[0676]
步骤-5:1-(苯磺酰基)-7-氯-6-(二氟甲基)吲哚xi-6的合成:
[0677]
向1-(苯磺酰基)-7-氯-吲哚-6-甲醛xi-6d(1.20g,3.1mmol)在dcm(30ml)中的溶液中加入氟化二乙基氨基硫(1.78ml,12.4mmol)在0℃,将该反应混合物在室温搅拌4h。通过tlc监测反应进程。完成后,将该反应混合物倾入冰h2o(50ml),用饱和nahco3溶液(40ml)碱化,用etoac(2
×
50ml)萃取。分离有机层,用盐水(50ml)洗涤,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,20%在己烷中的etoac),得到1-(苯磺酰基)-7-氯-6-(二氟甲基)吲哚xi-6(0.65g),为灰白色固体。
[0678]
收率:60%
[0679]
碱性lcms方法2(es-):340(m-h)-,97%纯度。
[0680]
b.7. 6-氯-7-(三氟甲氧基)-1h-吲哚xi-7的合成:
[0681][0682]
在-78℃向1-氯-3-硝基-2-(三氟甲氧基)苯(2.10g,8.69mmol)在thf(30ml)中的溶液中滴加溴化乙烯基镁(1m的thf溶液,34.8ml,34.8mmol),将该反应混合物在相同温度搅拌3h。将该反应混合物在室温再搅拌16h。通过tlc和lcms监测反应进程。完成后,用饱和
nh4cl(100ml)使该反应混合物淬灭,etoac(3
×
80ml)用萃取。分离有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,2-4%在己烷中的etoac),得到6-氯-7-(三氟甲氧基)-1h-吲哚xi-7(1.10g),为淡黄色液体。
[0683]
收率:49%。
[0684]
碱性lcms方法2(es-):234(m-h)-,92%纯度。
[0685]1h nmr(400mhz,dmso-d6)δ6.61(t,j=2.4hz,1h)7.15(d,j=8.0hz,1h)7.54(t,j=2.4hz,1h)7.62(d,j=8.8hz,1h)11.8(brs,1h)。
[0686]
c.式xii的中间体的合成
[0687]
c.1.方法a.6,7-二氯-1h-吲哚-3-磺酰氯xii-1的合成
[0688][0689]
在0℃向6,7-二氯-1h-吲哚xi-1(1.35g,6.89mmol)在ch3cn(15ml)中的溶液中滴加氯磺酸(1.60ml,24.1mmol),将该反应混合物在相同温度搅拌1h。将该反应混合物在室温搅拌2h。通过tlc和lcms监测反应进程。完成后,将该反应混合物倾入冰冷h2o(50ml),用etoac(3
×
50ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到6,7-二氯-1h-吲哚-3-磺酰氯xii-1(0.80g),为棕色固体。
[0690]
将该化合物原样用于下一步反应,未经进一步纯化。
[0691]
收率:39%。
[0692]
碱性lcms方法2(es-):282(m-h)-,50%纯度。
[0693]
可以根据与方法a类似的方法合成下表4中的中间体。
[0694]
表4:
[0695]
n
°
吲哚xi条件,时间收率(%)xii-2xi-2rt,2h25xii-3xi-3rt,2h粗制品xii-4xi-3art,2h粗制品xii-5xi-5rt,2h43xii-6xi-6rt,2h粗制品
[0696]
6-氯-7-环丙氧基-1h-吲哚-3-磺酰氯xii-2
[0697]
[0698]
碱性lcms方法2(es-):304(m-h)-,16%纯度。
[0699]
6-氯-7-(二氟甲基)-1h-吲哚-3-磺酰氯xii-3
[0700][0701]1h nmr(400mhz,cdcl3)δ7.33(t,j=54hz,1h),7.44-7.50(m,1h),8.08(d,j=2.93hz,1h),8.11(d,j=8.31hz,1h),9.51(brs,1h)。
[0702]
7-溴-6-氯-1h-吲哚-3-磺酰氯xii-4
[0703][0704]1h nmr(400mhz,dmso-d6)δ7.23(d,j=8.37hz,1h),7.41(d,j=2.46hz,1h),7.73(d,j=8.37hz,1h),11.50(brs,1h)。
[0705]
6-(二氟甲基)-7-氟-1h-吲哚-3-磺酰氯xii-5
[0706][0707]1h nmr(400mhz,dmso-d6)δ7.17-7.21(m,1h),7.29(t,j=54hz 1h),7.52(d,j=2.45hz,1h),7.66(d,j=8.31hz,1h),11.92(br s,1h)。
[0708]
7-氯-6-(二氟甲基)-1h-吲哚-3-磺酰氯xii-6
[0709][0710]
粗制品
[0711]
c.2. 1-(苯磺酰基)-6-氯-7-氟-吲哚-3-磺酰氯xii-7的合成
[0712]
步骤-1:1-(苯磺酰基)-6-氯-7-氟-吲哚xii-7a的合成
[0713]
向在冰浴上部预冷却的搅拌的氢氧化钠粉(3.54g,0.088mol)在二氯甲烷(60ml)中的混悬液中一次性加入6-氯-7-氟-1h-吲哚xi-4(5g,0.029mol),随后加入酸式硫酸四丁基铵(0.501g,0.001mol)。再持续搅拌10分钟,然后滴加苯磺酰氯(4.2ml,0.033mol)在二氯甲烷(15ml)中的溶液,历时20分钟,将该反应混合物在0℃搅拌1小时。除去冰浴,将该混合物在环境温度再搅拌1小时。用kieselguhr垫过滤该反应混合物,用二氯甲烷(2x 50ml)冲洗滤饼。用水(4x 50ml)和盐水(50ml)洗涤滤液,用无水硫酸钠干燥,过滤,真空浓缩溶剂,得到1-(苯磺酰基)-6-氯-7-氟-吲哚xii-7a(8.57g),为深褐色固体。
[0714]
收率:90%。
[0715]1h nmr(400mhz,dmso-d6)δ6.95(dd,j=3.7,2.3hz,1h),7.37(dd,j=8.4,6.2hz,1h),7.48(d,j=8.6hz,1h),7.66(tt,j=6.9,1.9hz,2h),7.80
–
7.71(m,1h),7.98
–
7.91(m,2h),7.99(d,j=3.7hz,1h)。
[0716]
步骤-2:1-(苯磺酰基)-6-氯-7-氟-吲哚-3-磺酰氯xii-7的合成
[0717]
向在冰浴上部预冷却的搅拌的1-(苯磺酰基)-6-氯-7-氟-吲哚xii-7a(8.50g,0.027mol)在乙腈(85ml)中的溶液中滴加氯磺酸(9.12ml,0.137mol),历时20min,将该反应混合物在环境温度搅拌16小时。将该反应混合物在搅拌下缓慢地倾入冰水(340ml),历时20分钟。通过过滤采集沉淀的固体,用冰水(3x 50ml)和环己烷(50ml)冲洗滤饼。然后在氮气流中干燥滤饼2小时,然后在40℃烘箱中干燥16小时,得到1-(苯磺酰基)-6-氯-7-氟-吲哚-3-磺酰氯xii-7(7.82g),为浅粉红色固体。
[0718]
收率:66%。
[0719]
酸性lcms方法4(es
+
):388(m+h)
+
,95%纯度。
[0720]1h nmr(400mhz,dmso-d6)δ7.44(dd,j=8.5,6.3hz,1h),7.72
–
7.61(m,3h),7.80
–
7.72(m,1h),7.81(s,1h),8.05
–
7.98(m,2h)。
[0721]
c.3. 6-氯-7-(三氟甲氧基)-1h-吲哚-3-磺酰氯xii-8的合成
[0722][0723]
步骤-1:6-氯-7-(三氟甲氧基)-1h-吲哚-3-磺酸xii-8a的合成:
[0724]
在0℃向6-氯-7-(三氟甲氧基)-1h-吲哚xi-7(0.10g,0.39mmol)在ch3cn(3ml)中的溶液中滴加clso3h(0.10ml),将该反应混合物在室温搅拌3h。通过tlc和lcms监测反应进程。完成后,将该反应混合物倾入冰冷h2o(25ml),用etoac(2
×
20ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到6-氯-7-(三氟甲氧基)-1h-吲哚-3-磺酸xii-8a(0.11g粗制品),为淡棕色液体。
[0725]
将该化合物原样用于下一步反应,未经进一步纯化。
[0726]
碱性lcms方法2(es-):314(m-h)-,91%纯度。
[0727]
步骤-2:6-氯-7-(三氟甲氧基)-1h-吲哚-3-磺酰氯xii-8的合成:
[0728]
在0℃向6-氯-7-(三氟甲氧基)-1h-吲哚-3-磺酸xii-8a(0.11g,0.28mmol)在ch3cn(3ml)中的溶液中加入pocl3(0.1ml,1.12mmol),将该反应混合物在60℃加热16h。通过tlc和lcms监测反应进程。完成后,真空浓缩该反应混合物。用冰冷h2o(50ml)稀释残余物,用etoac(3
×
40ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩。将得到的粗物质(0.1g,0.3mmol)溶于ch3cn(6ml),在0℃滴加pocl3(0.11ml,1.22mmol),将该反应混合物在60℃加热16h。完成后,真空浓缩该反应混合物。用稀释残余物冰冷h2o(60ml),用etoac(3
×
50ml)萃取。分离有机层,用无水na2so4干燥,真空浓缩,得到6-氯-7-(三氟甲氧基)-1h-吲哚-3-磺酰氯xii-8(0.13g粗制品),为棕色液体。
[0729]
将该化合物原样用于下一步反应,未经进一步纯化。
[0730]
碱性lcms方法2(es-):332(m-h)-[0731]
实施例化合物
[0732]
d.通式i化合物的合成
[0733]
本申请具体公开的所有本发明的化合物都被命名为“i-x”,其中任何“x”是指鉴别各个化合物的数字。因此,将实施例化合物命名为i-1,i-2,i-3等。这与任何化合物是否也可以用本申请的任何子通式例如式ii、iii或iv等进行描述无关。
[0734]
d.1.方法b.6,7-二氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺i-1的合成
[0735][0736]
在0℃向3,6-二氟-5-(2-氟乙氧基)吡啶-2-胺x-1(0.10g,0.44mmol)在吡啶(5ml)中的溶液中分批加入6,7-二氯-1h-吲哚-3-磺酰氯xii-1(0.33g,1.10mmol),历时20min期限,随后在相同温度添加dmap(0.01g,0.09mmol)。将该反应混合物在100℃加热30h。通过tlc和lcms监测反应进程。完成后,真空浓缩该反应混合物。将残余物与2n hcl(10ml)一起研磨,用h2o(20ml)稀释,用etoac(3
×
25ml)萃取。分离有机层,用盐水(2
×
30ml)洗涤,用无
水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,30%在己烷中的etoac),得到6,7-二氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺i-1(0.065g),为白色固体。
[0737]
收率:34%。
[0738]
碱性lcms方法2(es
+
):440(m+h)
+
,99%纯度。
[0739]1h nmr(400mhz,dmso-d6)δ4.22-4.25(m,1h),4.31-4.34(m,1h),4.62-4.65(m,1h),4.75-4.79(m,1h),7.39(d,j=8.80hz,1h),7.70-7.76(m,2h),7.97(s,1h),10.64(brs,1h),12.52(brs,1h)。
[0740]
可以根据与方法b类似的方法合成下表5中的化合物。
[0741]
表5:
[0742][0743][0744]
6-氯-7-环丙基氧基-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺i-2
[0745][0746]
碱性lcms方法2(es
+
):462(m+h)
+
,99%纯度。
[0747]1h nmr(400mhz,dmso-d6)δ0.54-0.56(m,2h),0.86-0.88(m,2h),4.25-4.27(m,1h),4.32-4.37(m,1h),4.40-4.43(m,1h),4.63-4.65(m,1h),4.75-4.78(m,1h),7.20(d,j=8.31hz,1h),7.48(d,j=8.80hz,1h),7.79(s,1h),7.92(s,1h),10.56(brs,1h),12.31(brs,1h)。
[0748]
6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-7-(二氟甲基)-1h-吲哚-3-磺酰胺i-3
[0749][0750]
碱性lcms方法2(es
+
):456(m+h)
+
,99%纯度。
[0751]1h nmr(400mhz,dmso-d6)δ4.27-4.32(m,1h),4.35-4.41(m,1h),4.64-4.69(m,1h),4.74-4.80(m,1h),7.38(d,j=8.86hz,1h),7.53(t,j=56hz,1h),7.83(t,j=9.11hz,1h),7.90-7.96(m,2h),10.65(s,1h),12.28(brs,1h)。
[0752]
7-溴-6-氯-n-[3,6-二氟-5-(2-氟乙氧基)-2-吡啶基]-1h-吲哚-3-磺酰胺i-4
[0753][0754]1h nmr(400mhz,dmso-d6)δ4.27-4.30(m,1h),4.34-4.39(m,1h),4.64-4.67(m,1h),4.76-4.80(m,1h),7.41(d,j=8.31hz,1h),7.75(d,j=8.80hz,1h),7.80-7.86(m,1h),7.97(d,j=2.93hz,1h),10.64(s,1h),12.47(brs,1h)。
[0755]
6,7-二氯-n-[3-氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺i-5
[0756][0757]
碱性lcms方法2(es
+
):422(m+h)
+
,99%纯度。
[0758]1h nmr(400mhz,dmso-d6)δ4.21-4.26(m,1h),4.30-4.34(m,1h),4.62-4.67(m,1h),4.74-4.79(m,1h),7.40(d,j=8.67hz,1h),7.52(dd,j=11.27,2.60hz,1h),7.71(d,j=8.67hz,1h),7.83(d,j=2.60hz,1h),7.97(s,1h),10.43(s,1h),12.60(br s,1h)。
[0759]
6,7-二氯-n-[5-(2,2-二氟乙氧基)-3-氟吡啶-2-基]-1h-吲哚-3-磺酰胺i-6
[0760][0761]
碱性lcms方法2(es
+
):440(m+h)
+
,99%纯度。
[0762]1h nmr(400mhz,dmso-d6)δ4.37(td,j=14.74,3.47hz,2h),6.22-6.52(m,1h),7.40(d,j=8.24hz,1h),7.58(dd,j=11.27,2.60hz,1h),7.73(d,j=8.67hz,1h),7.86(d,j=2.17hz,1h),7.98(s,1h),10.52(br s,1h),12.61(br s,1h)。
[0763]
6-氯-7-(二氟甲基)-n-[3-氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺i-7
[0764][0765]
碱性lcms方法2(es
+
):438(m+h)
+
,98%纯度。
[0766]1h nmr(400mhz,dmso-d6)δ4.20-4.26(m,1h),4.28-4.35(m,1h),4.60-4.66(m,1h),4.71-4.79(m,1h),7.35(d,j=8.80hz,1h),7.48-7.50(m,1h),7.52(t,j=52hz,1h),7.82(d,j=1.96hz,1h),7.87-7.93(m,2h),10.43(s,1h),12.21(br s,1h)。
[0767]
6-(二氟甲基)-7-氟-n-[3-氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺i-8
[0768][0769]
碱性lcms方法2(es
+
):422(m+h)
+
,98%纯度。
[0770]
7-氯-n-[3-氟-5-(2-氟乙氧基)吡啶-2-基]-6-(二氟甲基)-1h-吲哚-3-磺酰胺i-9
[0771][0772]
碱性lcms方法2(es
+
):456(m+h)
+
,98%纯度。
[0773]
6,7-二氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-基]-1h-吲哚-3-磺酰胺i-13
[0774][0775]
碱性lcms方法2(es
+
):470(m+h)
+
,99%纯度。
[0776]1h nmr(400mhz,dmso-d6)δ3.43(s,3h)4.28(td,j=14.43,3.42hz,2h)6.19-6.50(m,1h)7.39(d,j=8.80hz,1h)7.53(d,j=10.27hz,1h)7.71(d,j=8.31hz,1h)8.00(d,j=1.96hz,1h)10.36(brs,1h)12.58(brs,1h)。
[0777]
6-氯-n-[5-(2,2-二氟乙氧基)-3-氟吡啶-2-基]-7-(二氟甲基)-1h-吲哚-3-磺酰胺i-14
[0778][0779]
碱性lcms方法2(es
+
):456(m+h)
+
,99%纯度。
[0780]1h nmr(400mhz,dmso-d6)δ4.37(td,j=14.67,3.42hz,2h)6.22-6.52(m,1h)7.37(d,j=8.80hz,1h)7.53(t,j=54hz,1h)7.56-7.62(m,1h)7.87(d,j=1.96hz,1h)7.89-7.96(m,2h)10.54(s,1h)12.24(brs,1h)。
[0781]
6-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-基]-7-(二氟甲基)-1h-吲哚-3-磺酰胺i-15
[0782][0783]
碱性lcms方法2(es
+
):486(m+h)
+
,97%纯度。
[0784]1h nmr(400mhz,dmso-d6)δ3.41(s,3h)4.29(td,j=14.43,3.42hz,2h)6.19-6.50(m,1h)7.37(d,j=8.31hz,1h)7.52-7.57(m,1h)7.53(t,j=54hz,1h)7.90-7.95(m,2h)10.35(s,1h)12.21(brs,1h)。
[0785]
6,7-二氯-n-[5-(二氟甲氧基)-3-甲氧基吡啶-2-基]-1h-吲哚-3-磺酰胺i-16
[0786][0787]
碱性lcms方法2(es
+
):438(m+h)
+
,99%纯度。
[0788]1h nmr(400mhz,dmso-d6)δ3.78(s,3h)7.16(t,j=74hz,1h)7.25(d,j=2.45hz,1h)7.41(d,j=8.80hz,1h)7.62(d,j=1.96hz,1h)7.92(d,j=8.80hz,1h)8.09(s,1h)10.28(brs,1h)12.59(brs,1h)。
[0789]
6-氯-n-[5-(二氟甲氧基)-3-甲氧基吡啶-2-基]-7-(二氟甲基)-1h-吲哚-3-磺酰胺i-17
[0790][0791]
碱性lcms方法2(es
+
):454(m+h)
+
,99%纯度。
[0792]1h nmr(400mhz,dmso-d6)δ3.78(s,3h)7.16(t,j=74hz,1h)7.25(d,j=1.47hz,1h)7.39(s,1h)7.52(t,j=54hz,1h)7.62(d,j=1.47hz,1h)8.02(s,1h)8.13(d,j=8.31hz,1h)10.30(brs,1h)12.21(brs,1h)。
[0793]
6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-7-(三氟甲氧基)-1h-吲哚-3-磺酰胺i-18
[0794][0795]
碱性lcms方法2(es
+
):490(m+h)
+
,99%纯度。
[0796]1h nmr(400mhz,dmso-d6)δ4.23-4.25(m,1h)4.31-4.33(m,1h)4.61-4.67(m,1h)4.73-4.79(m,1h)7.36(d,j=8.31hz,1h)7.71-7.79(m,1h)7.83(d,j=8.80hz,1h)8.05(brs,1h)10.69(s,1h)12.70(brs,1h)。
[0797]
n-[5-(2,2-二氟乙氧基)-3-氟吡啶-2-基]-6-(二氟甲基)-7-氟-1h-吲哚-3-磺酰胺i-19
[0798][0799]
碱性lcms方法2(es
+
):440(m+h)
+
,82%纯度。
[0800]1h nmr(400mhz,dmso-d6)δ4.36(td,j=14.68,3.47hz,2h)6.22-6.51(m,1h)7.33(t,j=54hz,1h)7.33-7.38(m,1h)7.58(dd,j=11.10,2.31hz,1h)7.69(d,j=8.32hz,1h)7.86(d,j=2.31hz,1h)8.11(s,1h)10.54(brs,1h)12.92(brs,1h)。
[0801]
7-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-基]-6-(二氟甲基)-1h-吲哚-3-磺酰胺i-20
[0802][0803]
碱性lcms方法2(es
+
):486(m+h)
+
,98%纯度。
[0804]1h nmr(400mhz,dmso-d6)δ3.41(s,3h)4.28(td,j=14.55,3.18hz,2h)6.19-6.49(m,1h)7.33(t,j=56hz,1h)7.48(d,j=8.80hz,1h)7.54(d,j=10.27hz,1h)7.86(d,j=8.31hz,1h)8.12(s,1h)10.40(brs,1h)12.73(brs,1h)。
[0805]
6,7-二氯-n-[5-(2,2-二氟乙氧基)-3-甲氧基吡啶-2-基]-1h-吲哚-3-磺酰胺i-21
[0806][0807]
碱性lcms方法2(es
+
):452(m+h)
+
,98%纯度。
[0808]1h nmr(400mhz,dmso-d6)δ3.67(s,3h)4.29(td,j=14.67,2.93hz,2h)6.15-6.46(m,1h)7.04(d,j=1.47hz,1h)7.36(d,j=8.80hz,1h)7.48(s,1h)7.83(d,j=8.31hz,1h)7.96(s,1h)9.89(br s,1h)12.48(brs,1h)
[0809]
d.2. 6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺i-10的合成
[0810][0811]
向3,6-二氟-5-(2-氟乙氧基)吡啶-2-胺x-1(50mg,0.26mmol)在吡啶(1ml)中的溶液中加入1-(苯磺酰基)-6-氯-7-氟-吲哚-3-磺酰氯xii-7(106mg,0.26mmol),然后在室温搅拌3h。将该反应混合物蒸发至干,然后倾入水,用乙酸乙酯萃取(2次)。用mgso4干燥有机相,蒸发。通过柱色谱法纯化残余物(硅胶,100-200目,0-10%meoh的dcm溶液),然后进行碱性制备型lcms方法1,得到6mg 6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺i-10,为淡黄色固体。
[0812]
收率:5%。
[0813]
碱性lcms方法1(es-):422(m-h)-,97%纯度。
[0814]
d.3 6-氯-n-[5-(2,2-二氟乙氧基)-3,6-二氟吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺i-11的合成
[0815][0816]
向5-(2,2-二氟乙氧基)-3,6-二氟吡啶-2-胺x-4(154mg,0.37mmol)在吡啶(1ml)中的溶液中加入1-(苯磺酰基)-6-氯-7-氟-吲哚-3-磺酰氯xii-7(150mg,0.37mmol),然后在70℃搅拌2h。将该反应混合物蒸发至干,然后倾入水,用乙酸乙酯萃取(2次)。用mgso4干燥有机相,蒸发。通过柱色谱法纯化残余物(硅胶,100-200目,0-5%meoh的dcm溶液),然后进行碱性制备型lcms方法1,得到32mg 6-氯-n-[5-(2,2-二氟乙氧基)-3,6-二氟吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺i-11,为淡黄色固体。
[0817]
收率:20%。
[0818]
碱性lcms方法1(es-)
:440(m-h
)-,97%纯度。
[0819]1h nmr(400mhz,dmso-d6)δ4.41(td,j=14.6,3.5hz,2h),6.38(t,j=3.5hz,1h),7.31(dd,j=8.6,6.5hz,1h),7.58(d,j=8.6hz,1h),7.89(t,j=9.1hz,1h),8.06(d,j=2.4hz,1h),10.72(s,1h),12.84(s,1h)。
[0820]
d.4 6-氯-7-环丙基-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺i-12的合成
[0821][0822]
向7-溴-6-氯-n-[3,6-二氟-5-(2-氟乙氧基)-2-吡啶基]-1h-吲哚-3-磺酰胺i-4(0.25g,0.51mmol)在甲苯(8ml)和h2o(0.8ml)中的溶液中加入k3po4(0.22g,1.01mmol)、环丙基硼酸(0.07g,0.76mmol)和三环己基膦(0.03g,0.10mmol)。将该反应混合物用氩气吹扫15min,随后添加pd(oac)2(0.01g,0.05mmol)。将该反应混合物用氩气吹扫5min,在100℃加热16h。通过tlc和lcms监测反应进程。完成后,通过硅藻土垫过滤该反应混合物,用etoac(100ml)洗涤。用h2o(50ml)洗涤滤液。分离有机层,用无水na2so4干燥,真空浓缩。通过柱色谱法纯化得到的粗制品(硅胶,100-200目,20%在己烷中的etoac),得到6-氯-7-环丙基-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺i-12(0.04g),为灰白色固体。
[0823]
收率:17%。
[0824]
碱性lcms方法2(es
+
):446(m+h)
+
,95%纯度。
[0825]1h nmr(400mhz,dmso-d6)δ0.68-0.70(m,2h),1.09-1.17(m,2h),1.93-2.00(m,1h),4.25-4.29(m,1h),4.32-4.37(m,1h),4.62-4.67(m,1h),4.74-4.79(m,1h),7.19(d,j=8.80hz,1h),7.60(d,j=8.31hz,1h),7.77-7.84(m,1h),7.87(d,j=2.93hz,1h),10.55(s,1h),11.90(brs,1h)。
[0826]
d.5 6-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺i-22的合成
[0827][0828]
步骤-1:1-(苯磺酰基)-6-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基-2-吡啶基]-7-氟-吲哚-3-磺酰胺i-22a的合成
[0829]
在氩气气氛中,在密封试管中,将5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-胺x-5(54mg,0.24mmol)溶于吡啶(2ml)中。在0℃加入1-(苯磺酰基)-6-氯-7-氟-吲哚-3-磺酰氯xii-7(100mg,0.24mmol),然后在70℃搅拌过夜。将该反应混合物蒸发至干。通过硅胶急骤色谱法纯化残余物(用dcm和甲醇从100/0至98/2的梯度洗脱),得到96mg 1-(苯磺酰基)-6-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基-2-吡啶基]-7-氟-吲哚-3-磺酰胺i-22a,为棕色油状物。
[0830]
收率:66%。
[0831]
碱性lcms方法1(es-):592(m-h)-[0832]
步骤-2:n-[6-(二氟甲氧基)-5-氟-2-甲氧基吡啶-3-基]-6-(二氟甲基)-1h-吲哚-3-磺酰胺i-16的合成
[0833]
在密封试管中,将1-(苯磺酰基)-6-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基-2-吡啶基]-7-氟-吲哚-3-磺酰胺i-22a(96mg,0.16mmol)溶于thf(1ml)中。加入氟化四丁基铵(430mg,0.48mmol),将该反应混合物在70℃搅拌3天。将该反应混合物蒸发至干。将残余物溶于acoet,用水洗涤。用mgso4干燥有机相,蒸发。通过碱性制备型hplc(方法1)纯化残余物,得到27mg 6-氯-n-[5-(2,2-二氟乙氧基)-3-氟-6-甲氧基吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺i-22,为白色固体。
[0834]
收率:37%。
[0835]
碱性lcms方法1(es-):452(m-h)-,96%纯度。
[0836]1h nmr(400mhz,dmso-d6)δ3.45(s,3h),4.30(t,j=14.8hz,2h),6.36(s,1h),7.30
(t,j=7.6hz,1h),7.56(t,j=11.5hz,2h),8.06(s,1h),10.36(s,1h),12.78(s,1h)。
[0837]
d.6 6-氯-n-[5-[2-(二氟甲氧基)乙氧基]-3-氟-6-甲氧基吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺i-23的合成
[0838][0839]
向5-(2-(二氟甲氧基)乙氧基)-3-氟-6-甲氧基吡啶-2-胺x-8(74mg,0.29mmol)在吡啶(2ml)中的溶液中加入1-(苯磺酰基)-6-氯-7-氟-吲哚-3-磺酰氯xii-7(100mg,0.24mmol),然后在70℃搅拌2h。将该反应混合物蒸发至干,然后通过碱性制备型lcms方法1纯化残余物,得到54mg 6-氯-n-[5-[2-(二氟甲氧基)乙氧基]-3-氟-6-甲氧基吡啶-2-基]-7-氟-1h-吲哚-3-磺酰胺i-23,为淡粉红色固体。
[0840]
收率:45%。
[0841]
碱性lcms方法1(es-)482(m-h)-,99%纯度。
[0842]1h nmr(400mhz,dmso-d6)δ3.40(s,3h),4.21
–
4.03(m,4h),6.71(s,1h),7.29(dd,j=8.6,6.5hz,1h),7.56(d,j=8.6hz,1h),7.45(d,j=10.3hz,1h),8.03(s,1h),nh质子不可见。
[0843]
对比例1的合成
[0844]
对比例1:6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺
[0845][0846]
对比例1
t-rex cho细胞(cho hgpr17)在37℃在5%co2的潮湿气氛中进行培养。细胞在含有补充有潮霉素b(500μg/ml)和杀稻瘟素(30μg/ml)的营养混合物f-12的dmem中生长。通过在测定前用多西环素(1μg/ml)处理16-20小时来诱导flp-in基因座的表达。
[0865]
原代少突神经胶质细胞:
[0866]
在出生后第0天至第2天从wistar大鼠幼仔的前脑分离出原代少突神经胶质细胞祖细胞(opc)。用注射器和两个不同的空心针(先用1.2
×
40,然后用0.60
×
30)机械解离脑。将无团块的细胞悬浮液通过70-μm细胞过滤器过滤,并接种到dmem中的聚-d-赖氨酸包被的75-cm2培养瓶中,所述dmem补充有10%(v/v)热灭活的胎牛血清,青霉素(100单位/ml)和链霉素(0.1mg/ml),每隔一天更换一次培养基。在37℃,5%co2的潮湿气氛中8至11天后,将混合的培养物在240rpm下摇动14-24小时,以从星形胶质细胞和小胶质细胞中分离opc。为了进一步富集opc,将悬浮液接种在未涂布的培养皿上45分钟。然后,将opc接种到聚l-鸟氨酸包被的平板中,并在增殖的neurobasal培养基中在5%co2的潮湿气氛中维持在37℃,所述neurobasal培养基补充有2%(v/v)b27,2mm glutamax,100单位/ml青霉素,0.1mg/ml链霉素,10ng/ml pdgf-aa和10ng/ml碱性fgf,每隔一天更换一次培养基。
[0867]
b-ii:功能性体外gpr17测定法
[0868]
b-ii-a:钙动员功能测定法
[0869]
gpr17是g蛋白偶联受体。gpr17激活触发gq型g蛋白信号传导,导致内质网钙(ca
2+
)储存在胞质溶胶中,其可以使用钙5染料(细胞溶质ca
2+
水平的荧光指示染料)测量。在ca
2+
测定或在gpr17camp测定中评估本发明的化合物,如下文进一步描述。如下表5所示,在两个活性测试中都测定了一些代表性实施例。
[0870]
ca
2+
测定法的描述:
[0871]
将cho hgpr17解冻并以每孔20,000个细胞的密度接种到具有透明底部的黑色384孔板中。将细胞在37℃,5%co2的潮湿气氛中培养过夜。接种后16至20小时,根据制造商的说明,用钙5染料(细胞溶质ca
2+
指示剂荧光染料)加载cho hgpr17 60分钟。在室温在flipr tetra读数器中随时间记录相对于细胞溶质ca
2+
浓度的荧光信号。首先将细胞在室温在含有递增浓度的测试化合物(通常10-11
m至10-6
m)的hbss hepes缓冲液ph 7.4中温育30分钟。然后,将50nm mdl29,951(一种gpr17激动剂)加入到细胞中。测量不同浓度的测试化合物的抑制作用并测定所得的pic
50
。所有温育均一式两份进行,并将结果与gpr17激动剂和拮抗剂参照化合物的浓度响应曲线进行比较。使用xlfit 4-参数逻辑方程y=a+((b-a)/(1+((c/x)^d)))在activitybase xe中进行分析和曲线拟合,其中a,b,c和d代表最小值y,最大值y,ic
50
和斜率。
[0872]
ca
2+
测定法的结果:
[0873]
当在ca
2+
动员测定法中测试时,实施例的化合物典型地表现出大于或等于6.5的pic
50
值;更优选大于或等于7.5,甚至更优选大于或等于8.5。所测试的实施例化合物的活性描述于下面b2b部分的表中。如下所示活性范围a、b和c是指ca
2+
测定法中的pic
50
值:“a”:pic
50
<7.5,“b”:pic
50 7.5≤x<8.5,“c”:8.5≤pic
50
。
[0874]
b-iib.camp累积功能测定法
[0875]
gpr17活化还可以募集gi型g蛋白信号传导,导致细胞内环磷酸腺苷(camp)的减少。可以使用来自cisbio(codolet,法国)的htrf camp动态测定套装产品测量细胞内camp
变化。使用同源时间分辨荧光技术(htrf),该测定基于细胞产生的天然camp和用染料d2标记的camp之间的竞争。示踪剂结合由用密码子标记的抗camp抗体确定。
[0876]
camp测定法的描述
[0877]
用含有edta的pbs分离cho hgpr17,并以每孔5,000个细胞在黑色384孔板中分配。首先将细胞在室温在含有介质或不同浓度的测试gpr17拮抗剂/反向激动剂化合物的hbss hepes(ph7.4)中温育30分钟。然后,将mdl29,951gpr17激动剂(通常为10-5
m至10-10
m)的剂量响应曲线加入介质和每种测试gpr17拮抗剂/反向激动剂化合物浓度,最终体积为20μl hbss hepes缓冲液(ph 7.4),含有1%dmso,5μm福司可林(forskolin)和0.1mm ibmx。在室温温育60分钟后,终止反应并根据制造商的说明通过将d2检测试剂和穴状化合物试剂各自加入10μl裂解缓冲液中来裂解细胞。温育60分钟后,根据制造商的说明使用具有激光激发的envision读板器测量camp浓度的变化。所有温育均一式两份进行。使用graphpad prism软件使用4-参数逻辑方程分析数据,以在不存在和存在gpr17拮抗剂/反向激动剂测试化合物的情况下测量mdl29,951pec
50
。将剂量比(dr)对抗拮抗剂浓度作图,并且schild分析提供gpr17拮抗剂/反向激动剂测试化合物的估计的亲和力pa2。
[0878]
camp测定法的结果:
[0879]
当在camp测定法中测试时,实施例的化合物典型地表现出大于或等于6.5的pa2值;优选大于或等于7.5;更优选大于或等于8.5。所测试的实施例化合物的活性描述于下表中。如下所示活性范围a、b和c是指camp测定法中的pa2值:“a”:pa2<7.5,“b”:pa
2 7.5≤x<8.5,“c”:8.5≤pa2。
[0880]
下表6显示了在ca
2+
和camp测定法中测试的实施例化合物的pic
50
和pa2值。pa2列中的空白表示相应的化合物还没有被测试,或结果还不可得到。
[0881]
表6:
[0882][0883]
b-iic:少突神经胶质细胞成熟/髓鞘化测定法
[0884]
gpr17的负性调节剂对原代少突神经胶质细胞成熟/髓鞘化的作用可以通过使用针对髓磷脂碱性蛋白(mbp)的抗体的免疫测定在体外评估,作为成熟少突神经胶质细胞的标记。
[0885]
mbp蛋白质印迹/少突神经胶质细胞/髓鞘化测定法的描述
[0886]
在增殖培养基中3-4天后,将大鼠原代opc以25,000个细胞/cm2接种于12孔组织培养板中,并转换为无生长因子的neurobasal培养基以诱导自发体外分化和gpr17蛋白表达。对于蛋白质表达的终末分化和定量分析,24-48小时后,无生长因子培养基补充0.20ng/ml三碘甲状腺原氨酸(t3)和10ng/ml睫状神经营养因子以及1μm gpr17拮抗剂/反向激动剂测试化合物或介质额外3天。化合物处理后,用冰冷的pbs洗涤细胞两次,并在冰冷的补充有蛋白酶抑制剂混合物的裂解缓冲液(25mm tris,ph 7.4,150mm nacl,1mm edta,1%triton x-100,1%igepal)中裂解。将裂解物在4℃旋转20分钟,并在4℃以15,000
×
g离心10分钟。根据制造商的说明,使用pierce bca蛋白质测定法测定蛋白质浓度。通过10%sds-聚丙烯酰胺凝胶电泳分离7.5-15μg蛋白质,并通过电印迹转移到硝酸纤维素膜上。洗涤后,在室温用roti-block封闭膜1小时,并在4℃在roti-block中用mbp抗体(1:5000,lifespan biosciences)温育过夜。用含有0.1%吐温的pbs洗涤膜3次,然后在室温用roti-block中的辣根过氧化物酶缀合的山羊抗小鼠igg抗体温育1小时。使用amersham biosciences ecl prime蛋白质印迹检测试剂通过化学发光使免疫反应性蛋白质可视化,并使用gelscan软件通过光密度测定法定量。为了标准化相同的加载和蛋白质转移,用抗-β-肌动蛋白的抗体(1:2500,biolegend;二抗山羊抗兔igg抗体hrp(abin))重新探测膜。将测试化合物存在下mbp表达水平的变化与对照条件下的mbp表达进行比较。
[0887]
mbp纤维板/少突神经胶质细胞成熟/髓鞘化测定法的描述
[0888]
在mimetix aligned 96孔纤维板(electrospining公司)中以16,000-22,000个细胞/cm2接种opc。在增殖培养基中2天和在无生长因子的neurobasal培养基中培养2天以诱导自发体外分化和gpr17蛋白表达之后,将介质或1μm拮抗剂/反向激动剂测试化合物加入补充有0.20ng/ml三碘甲状腺原氨酸和10ng/ml睫状神经营养因子的终末分化培养基中6天,3天后更换培养基。然后将细胞在4%多聚甲醛中固定,然后用pbs洗涤,在pbs中的0.1%tritonx-100中透化,并用磷酸盐缓冲盐水中的10%山羊血清和1%牛血清白蛋白封闭。将mbp抗体在封闭缓冲液(1:2000)中稀释,并在37℃温育1小时。将细胞再次在pbs中洗涤,并与针对小鼠igg的cy2-缀合的第二抗体(millipore,1:500)温育1小时。pbs洗涤后,将细胞用0.2μg/ml dapi染色,再次洗涤并用mowiol固定。使用具有apotome成像系统和plan-apochromat 20x/0.8物镜的zeiss axioobserver.z1显微镜拍摄荧光图像,其具有egfp滤光片(激发470/40nm;发射525/50nm)和dapi滤光片(激发365nm;发射445/50纳米)。使用用zeiss zen2.3软件处理的相同设置对至少15个用于对照(具有0.1%dmso的末端分化培养基)和用于测试化合物的随机区域进行成像。在不存在或存在gpr17负性调节剂的情况下,通过纤维长度组(0至40μm,41至60μm,61至80,81至100,101至120和>120μm)报告髓鞘化纤维数量的变化。
[0889]
cyp 450-1a2诱导测定法
[0890]
将来自单个供体的冷冻保存的人肝细胞接种在96孔胶原蛋白包被的板上,以使最终接种密度为0.1x 106细胞/孔(每孔的最终体积为0.1ml)。然后将细胞在37℃,95%湿度,5%co2的接种培养基中温育,以使细胞附着。4h后,将接种培养基替换为0.1ml的预温热无血清williams e培养基,其中包含100iu/ml青霉素,100μg/ml链霉素,10μg/ml胰岛素,2mm l谷氨酰胺和0.1μm氢化可的松。
[0891]
第二天,在测定培养基中向细胞中给予测试化合物(最终测试化合物浓度为10μm;
dmso最终浓度为0.1%)。将阳性对照诱导剂,对于cyp1a2为奥美拉唑(50μm),对于cyp2b6为苯巴比妥(500μm)和对于cyp3a4为利福平(10μm)与测试化合物一起温育。包括阴性对照孔,其中用介质溶剂(测定介质中的0.1%dmso)替代测试化合物。每个测试化合物一式三份给予。将细胞暴露于溶液中72h,每24h添加一次新鲜溶液。
[0892]
为了确定催化活性,在预温热的测定介质中制备探针底物,非那西丁(终浓度25μm),安非他酮(终浓度100μm)和咪达唑仑(终浓度2.5μm)的溶液。在72h暴露期结束时,用探针底物混合物替代培养基。将肝细胞温育30min。在温育期结束时取出等分试样,并以1:2的比例放入含有内标的甲醇中。将样品在4℃以2500rpm离心20min。用去离子水稀释上清液的等分试样,并使用cyprotex通用lc ms/ms方法对乙酰氨基酚、羟基安非他酮和1羟基咪达唑仑的水平进行定量。
[0893]
在室温,将肝细胞溶解在0.1m氢氧化钠中,并使用pierce
tm bca蛋白质测定套装产品(thermo scientific),应用牛血清白蛋白为标准品测定每个孔中的蛋白质含量。
[0894]
为了测定催化活性,将每个测试化合物重复样品形成的代谢物的浓度转换为cyp活性,并对蛋白质校准。通过与介质对照空进行比较来确定倍数改变。将在每种测试化合物浓度下观察到的倍数改变表示为每种p450同工型阳性对照化合物的百分比。
[0895]
与在r7位上具有氢的对比化合物相比,本发明的化合物典型地显示没有cyp 1a2诱导作用或显示至少显著更少的cyp 1a2诱导作用。
[0896]
作为举例说明,与介质对照相比,其中r7分别为氯、环丙基氧基、二氟甲基和环丙基的实施例化合物i-1、i-2、i-3和i-12显示没有cyp1a2诱导作用,而相应的其中r7为氢的对比例1(6-氯-n-[3,6-二氟-5-(2-氟乙氧基)吡啶-2-基]-1h-吲哚-3-磺酰胺)表现出16倍的cyp1a2诱导作用。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签:

 热门咨询
热门咨询




tips