
 商标分类
商标分类  商标转让
商标转让 
核酸扩增方法与流程
 2021-02-02 05:02:57|
2021-02-02 05:02:57| 245|
245| 起点商标网
起点商标网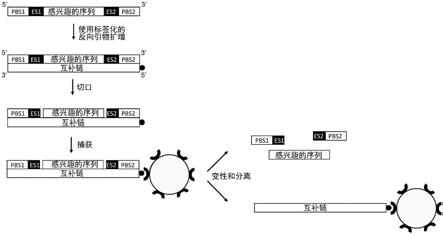
mj等人,nucleic acids res.2004;32(4):e47)描述了在该领域所做的改进。
[0007]
随着诸如janitz ed.next generation genome sequencing,wiley vch,2008中所描述的并可在由roche(gs flx和相关系统)和lllumina(基因组分析仪和相关系统)提供的平台市场上获得的新一代测序(next generation sequencing,ngs)技术的兴起,出现了使ola测定适应作为检测平台的测序的需要。该领域的改进尤其在keygene nv的wo 2007100243中进行了描述。在wo2007100243中,已经描述了新一代测序技术在寡核苷酸连接测定的结果中的应用。不仅从可靠性和准确性的角度,而且从经济驱动因素的角度来看,在该领域仍然需要进一步的改进,以通过扩大规模来进一步降低成本。
[0008]
例如,持续需要经济地生产高质量的寡核苷酸探针。此类高质量的寡核苷酸尤其适合用于多重反应,诸如本文中上文所描述的多重ola测定。ola测定通常需要三种特异性探针来指定每个靶点。在高度多重化的情况下,因为寡核苷酸通常是单独合成和纯化的,所以所需寡核苷酸的数目和量潜在地非常昂贵。porreca已经在2007年(porreca等人multiplex amplification of large sets of human exons,nature methods-4,931-936(2007))解决了此问题,并且公开了一种多重寡核苷酸探针(100-mers)的扩增方法,该多重寡核苷酸探针是在用于核酸靶向扩增方法的固体表面上并行合成的。porreca等人描述了一种使用pcr扩增探针的方法,这些探针各自包含人类基因组中的70nt连续蛋白编码序列,该连续蛋白编码序列侧接有含有识别位点的序列,这些识别位点用于在它们与靶向臂连接处的切口(nicking)限制性核酸内切酶。将扩增子使用res消化、柱纯化、在丙烯酰胺凝胶上分离、从对应于预期的单链70nt种类的条带中回收并纯化。根据该论文,该方法产生将200μl中的2.5nm寡核苷酸(即,0.5pmol的量)扩增为20μl中的125nm寡核苷酸(即,2.5pmol的量)。换句话说,报告了5倍的扩增。
[0009]
本发明人对porreca的探针扩增方法进行了重新设计,并且当使用相对大量的平均长度为90nt(85-93nt)的九种探针前体的输入材料(0.5pmol)时,发现了类似的结果,即,扩增系数(amplification factor)为4.5。对于使用高通量靶向核苷酸检测(诸如ola)来说,这种产量并不令人满意。进一步地,尽管3重测定(适合于3个不同靶序列的snp检测,并且需要9个不同的探针序列)产生了相对干净的扩增产物,但是将探针数目增加到326重测定(978个不同的探针序列)产生了背景条带,这可能是由于异源双链体形成而导致的,所述异源双链体形成可能会由于pcr扩增伪像(artifact)而阻碍产量和序列组成。
[0010]
因此,在本领域中,仍然需要一种方法来增加合并的寡核苷酸(例如在阵列上低量合成的)的摩尔量和/或产量,而且不会改变它们的序列组成和显著地干扰库中的每个寡核苷酸的相对丰度。需要以足够多的数量和足够好的质量生产这些寡核苷酸,以允许开发用于对数千个样本进行高通量分析的高度多重化测定。
[0011]
本发明人现在发现了一种实现高产量的改进的寡核苷酸扩增方法,即,在纯化后,甚至对于适合于高通量检测方法的326重测定也产生500倍的扩增系数。本发明在整个说明书、附图和本文描述的各种实施方案中进行了更详细地阐述。引用的所有参考文献均并入本文。
技术实现要素:
[0012]
在第一方面,本发明涉及一种用于生产具有感兴趣的序列的一个或多个单链寡核
苷酸的方法,其中方法包含以下步骤:
[0013]
a)提供至少一个单链或双链核酸前体,其包含第一链和任选地包含与第一链互补的第二链,其中第一链在5'至3'方向上包含以下元件:
[0014]
(1)第一引物结合位点;
[0015]
(2)第一核酸内切酶识别位点;
[0016]
(3)所述感兴趣的序列;
[0017]
(4)第二核酸内切酶识别位点;以及
[0018]
(5)第二引物结合位点;
[0019]
其中第一核酸内切酶识别位点被设计成使得在双链化(duplexing)之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架;并且,
[0020]
其中第二核酸内切酶识别位点被设计成使得在双链化之后,第二核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架;
[0021]
b)使用能够与第一引物结合位点杂交的第一引物以及能够与第二引物结合位点杂交的第二引物,通过扩增方法来扩增步骤a)的前体;
[0022]
c)用第一核酸内切酶和第二核酸内切酶消化在步骤b)中获得的扩增的双链前体,以通过在感兴趣的序列的紧接上游和下游切割糖-磷酸骨架来生产扩增的双链核酸前体;以及
[0023]
e)使扩增的双链前体变性,从而释放具有感兴趣的序列的单链寡核苷酸。
[0024]
优选地,第一引物能够选择性地仅与第一引物结合位点退火(更具体地,与包含在第二链的第一引物结合位点内的引物结合序列退火),且第二引物能够选择性地仅与第二引物结合位点退火(更具体地,与包含在第一链的第二引物结合位点内的引物结合序列退火)。任选地,第一引物可以与第一引物结合位点和第二引物结合位点二者退火,并且第二引物可以与第一引物结合位点和第二引物结合位点二者退火,从这个意义上来说,第一引物和第二引物可以相同或相似。任选地,该引物能够选择性地仅与第一引物结合位点和第二引物结合位点退火。
[0025]
优选地,感兴趣的序列不包含第一核酸内切酶识别位点和/或第二核酸内切酶识别位点或它们的反向互补。
[0026]
在优选的实施方案中,本发明的方法进一步包含一个或多个步骤,以便将包含与感兴趣的序列互补的序列的寡核苷酸与第一链、或与第一链的包含感兴趣的序列的其余部分分离。优选地,这通过在以下各项中添加使第二链、或第二链的至少包含与感兴趣的序列互补的序列的其余部分固定的步骤d)来实现:
[0027]
i)在扩增步骤b)与消化步骤c)之间,
[0028]
ii)在消化步骤c)与变性步骤e)之间;或者,
[0029]
iii)在变性步骤e)之后。
[0030]
优选地,该固定步骤涉及将第二链、或其包含与感兴趣的序列互补的序列的部分亲和捕获在固体支持物上。这可能需要对第二链的整体、或其包含与感兴趣的序列互补的序列的部分进行标签化。可以使用包含亲和标签的、本发明方法步骤b)中的第二引物来实现对第二链的整体进行标签化。亲和标签能够至少存在于第二引物上。在本文中应进一步理解,亲和标签可以存在于第一引物和第二引物二者上。可选地,亲和标签仅存在于第二引
物上,即,它不存在于第一引物上。第一引物和第二引物用于生产包含标签的扩增的双链核酸前体。可选地,在扩增之前,步骤b)中使用的第二引物可以存在于固体支持物上,其中步骤b)中的扩增在固体支持物上进行,从而产生通过第二链附接到固体支持物的扩增子。向本发明方法添加去除第二链、或其包含感兴趣的序列的反向互补的部分的进一步步骤,以获得具有感兴趣的序列的单链寡核苷酸。优选地,在如上文中所定义的选项i)或ii)中的变性步骤之后、或在如上文中所定义的选项iii)中的固定步骤之后,添加所述去除步骤。优选地,在该实施方案内,设计前体或方法,由此使得对如本发明方法步骤c)中所定义的扩增的双链前体的消化将保持始自标签并包括感兴趣的序列的第二链的糖-磷酸骨架完整。
[0031]
优选地,本发明方法进一步包含纯化单链寡核苷酸的步骤g)。
[0032]
在优选的实施方案中,步骤e)中的变性包括化学变性,其中优选地,化学变性是通过添加强碱、优选地通过添加浓度为约0.5-1.5m的碱金属氢氧化物来增加ph值而进行的。
[0033]
优选地,核酸前体由约20-200个核苷酸组成,并且其中优选地,核酸前体具有选自seq id no:1-seq id no:978的序列。
[0034]
优选地,感兴趣的序列至少部分地与预定基因组序列互补,其中优选地,生产的寡核苷酸适用于多重ola测定,并且其中更优选地,生产的寡核苷酸适用于至少300-重ola测定。
[0035]
优选地,核酸前体是单链核酸前体。
[0036]
在优选的实施方案中,步骤b)中的扩增方法是等温扩增方法,其中优选地,等温扩增方法是重组酶聚合酶扩增(recombinase polymerase amplification,rpa)或解旋酶依赖性扩增(helicase dependent amplification,hda)。
[0037]
优选地,步骤c)中的第一核酸内切酶和第二核酸内切酶是两种不同的酶。
[0038]
优选地,步骤c)中的第一核酸内切酶切割:i)第一dna链;ii)第一dna链和第二dna链。
[0039]
在优选的实施方案中,在结合步骤d)中的固体支持物之前,纯化来自步骤b)的扩增的双链前体。
[0040]
优选地,用于亲和捕获第二链或其部分的标签是生物素,并且固体支持物包含抗生蛋白链菌素,其中优选地,固体支持物是珠,并且其中更优选地,珠是磁性珠。
[0041]
优选地,在步骤a)中提供具有不同感兴趣的序列的两个或更多个核酸前体,其中优选地,核酸前体的序列选自seq id no:1-seq id no:978。
[0042]
在第二方面,本发明涉及一种单链或双链核酸前体,其包含第一链和任选地包含与第一链互补的第二链,其中所述第一链在5'至3'方向上包含以下元件:
[0043]
(1)第一引物结合位点;
[0044]
(2)第一核酸内切酶识别位点;
[0045]
(3)感兴趣的序列;
[0046]
(4)第二核酸内切酶识别位点;以及
[0047]
(5)第二引物结合位点;
[0048]
其中第一引物能够选择性地仅与第一引物结合位点退火,并且第二引物能够选择性地仅与第二引物结合位点退火;
[0049]
其中感兴趣的序列不包含第一核酸内切酶识别位点和第二核酸内切酶识别位点
或它们的反向互补;
[0050]
其中第一核酸内切酶识别位点被设计成使得在双链化之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架;并且,
[0051]
其中第二核酸内切酶识别位点被设计成使得在双链化之后,第二核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架。
[0052]
优选地,前体进一步包含位于第二链的5'端处的亲和标签,优选地,亲和标签不位于第一链的5'端处,优选地,亲和标签仅位于第二链的5'端处。
[0053]
在第三方面,本发明涉及通过亲和-捕获结合到固体支持物的如本文中所定义的双链核酸前体。
[0054]
在第四方面,本发明涉及一种试剂盒套装(kit of parts),其用于本发明方法,所述试剂盒套装包含:
[0055]-容器,其包含第二核酸内切酶和任选地包含第一核酸内切酶的;
[0056]-容器,其包含用于第一方面的方法中的扩增步骤b)的酶,任选地与第一和/或标签化的第二引物组合;
[0057]-容器,其包含用于亲和纯化的固体支持物;以及任选地
[0058]-容器,其包含用于变性的化学制品。
[0059]
在第五方面,本发明涉及如本文中所定义的核酸前体或如本文中所定义的试剂盒套装用于生产一个或多个单链寡核苷酸的用途。
[0060]
定义
[0061]
在整个说明书和权利要求书中使用了与本发明的方法、组合物、用途和其他方面有关的各种术语。除非另外指出,否则此类术语应被赋予其在本发明所属领域中的普通含义。其他特定定义的术语将以与本文中提供的定义一致的方式解释。尽管在实践中可以使用与本文描述的那些类似或等价的任何方法和材料来测试本发明,但是本文描述了优选的材料和方法。
[0062]
除非上下文清楚地另有指示,否则单数术语“一个”、“一种”和“该”包括复数指示物。因此,例如,对“一个细胞”的引用包括两个或更多个细胞的组合等。
[0063]
术语“和/或”是指这样的情形,其中一种或多种所述的情况可以单独发生或与至少一种所述情况组合发生、直到与所有所述情况组合发生。
[0064]
如本文所用,术语“约”用于描述和说明小的变化。例如,术语可以指小于或等于
±
(+或-)10%,诸如小于或等于
±
5%、小于或等于
±
4%、小于或等于
±
3%、小于或等于
±
2%、小于或等于
±
1%、小于或等于
±
0.5%、小于或等于
±
0.1%、或者小于或等于
±
0.05%。另外,数量、比率和其他数值在本文中有时是以范围形式呈现的。应当理解,此种范围形式是为了方便和简洁而使用的,应灵活地理解为包括明确指定为范围界限值的数值,而且包括范围内涵盖的所有单个数值或子范围,犹如每个数值和子范围均已明确指定。例如,在约1至约200的范围内的比率应理解为包括明确列举的约1和约200的界限值,而且包括诸如约2、约3和约4的单个比率以及诸如约10至约50、约20至约100等的子范围。
[0065]
术语“包含”被解释为是包括性的和开放式的,而不是排他性的。具体地,术语及其变化意味着包括指定的特征、步骤或组分。这些术语不应解释为排除其他特征、步骤或组分的存在。
[0066]“构建体”或“核酸构建体”或“载体”:这是指通过使用重组dna技术而产生的人造核酸分子,其用于将外源dna递送到宿主细胞中,通常是出于在宿主细胞中表达包含在构建体上的dna区域的目的。构建体的载体骨架可以是例如整合有(嵌合)基因的质粒,或者,如果已经存在合适的转录调节序列(例如,(诱导型)启动子),则仅将期望的核苷酸序列(例如,编码序列)整合在转录调节序列的下游。载体可以包含其他基因元件,以促进载体在分子克隆中的用途,诸如例如,可选择标志物、多个克隆位点等。
[0067]“序列”或“核苷酸序列”:这是指核酸的核苷酸的顺序或在核酸内的核苷酸的顺序。换句话说,核酸中核苷酸的任何顺序可以被称为序列或核苷酸序列。
[0068]
术语“同源性”、“序列相同性”等在本文中可互换地使用。序列相同性在本文中定义为两个或更多个氨基酸(多肽或蛋白质)序列或者两个或更多个核酸(多核苷酸)序列之间的关系,如通过对序列进行比较来确定。在本领域中,"相同性"还意指氨基酸或核酸序列之间的序列相关性程度,视情况而定,如通过这些序列的字符串之间的匹配确定。通过将一个多肽的氨基酸序列及其保守的氨基酸取代物与第二多肽的序列进行比较来确定两个氨基酸序列之间的“相似性”。
[0069]
术语“互补性”在本文中定义为序列与完全互补链(在本文下文中定义,例如,第二链)的序列相同性。例如,100%互补(或完全互补)的序列在本文中理解为与互补链具有100%序列相同性,并且例如,80%互补的序列在本文中理解为与(完全)互补链具有80%序列相同性。
[0070]
可以通过已知方法容易地计算“相同性”和“相似性”。取决于两个序列的长度,可以使用全局或局部比对算法通过对两个肽或两个核苷酸序列进行比对来确定“序列相同性”和“序列相似性”。长度相似的序列优选地使用全局比对算法(例如,needleman-wunsch比对)进行比对,全局比对算法在整体长度上对序列进行最佳比对,而长度相差很大的序列优选地使用局部比对算法(例如,smith waterman)进行比对。当序列(当通过例如使用默认参数的程序gap或bestfit进行最佳比对时)至少共享某一最小百分比的序列相同性(如下文中所定义的)时,它们可以被称为“基本相同”或“基本相似”。gap使用needleman和wunsch全局比对算法在两个序列的整体长度(全长)上进行比对,从而使匹配数最大化并且使空位数最小化。当两个序列具有相似的长度时,全局比对适合用于确定序列相同性。通常,使用gap默认参数,其中空位产生罚分=50(核苷酸)/8(蛋白质),空位延伸罚分=3(核苷酸)/2(蛋白质)。对于核苷酸,使用的默认得分矩阵是nwsgapdna,并且对于蛋白质,使用的默认得分矩阵是blosum62(henikoff&henikoff,1992,pnas89,915-919)。序列比对和序列相同性百分比得分可以使用计算机程序来确定,诸如gcg wisconsin package,10.3版(可获自accelrys inc.,9685scranton road,san diego,ca 92121-3752usa),或使用开源软件,诸如embosswin2.10.0版中的程序“needle”(使用全局needleman-wunsch算法)或“water”(使用局部smith waterman算法),使用与用于上文中gap相同的参数,或使用默认设置(对于“needle”和“water”二者并且对于蛋白质和dna比对二者,默认的空位开放罚分都是10.0,并且默认的空位延伸罚分都是0.5;对于蛋白质,默认得分矩阵是blosum62,并且对于dna,默认得分矩阵是dnafull。当序列的总长度相差很大时,优选局部比对,诸如使用smith waterman算法的那些。
[0071]
可选地,可以使用诸如fasta、blast等的算法通过检索公共数据库来确定相似性
或相同性百分比。因此,本发明的核酸序列和蛋白质序列可以进一步用作“查询序列”以执行针对公共数据库的检索来例如鉴定其他家族成员或相关序列。可以使用altschul等人(1990)j.mol.biol.215:403-10的blastn和blastx程序(2.0版)来进行此类检索。可以用nblast程序(分数=100,字长=12)来进行blast核苷酸检索,以得到与本发明的核酸分子同源的核苷酸序列。可以用blastx程序(分数=50,字长=3)来进行blast蛋白质检索,以得到与本发明的蛋白质分子同源的氨基酸序列。为了得到用于比较目的空位比对,可以采用如在altschul等人,(1997)nucleic acids res.25(17):3389-3402中所描述的gapped blast。当采用blast和gapped blast程序时,可以使用各自程序(例如,blastx和blastn)的默认参数。参见national center for biotechnology information的主页http://www.ncbi.nlm.nih.gov/.
[0072]
如本文所用,术语“选择性杂交”、“选择性地杂交”和类似术语旨在描述杂交和洗涤的条件,在该条件下,至少66%、至少70%、至少75%、至少80%、更优选至少85%、甚至更优选至少90%、优选至少95%、更优选至少98%或更优选至少99%彼此同源的核苷酸序列通常保持彼此杂交。也就是说,此类杂交序列可以共享至少45%、至少50%、至少55%、至少60%、至少65、至少70%、至少75%、至少80%、更优选至少85%、甚至更优选至少90%、更优选至少95%、更优选至少98%或更优选至少99%的序列相同性。
[0073]
此类杂交条件的优选非限制性实例是:在约45℃于6x氯化钠/柠檬酸钠(ssc)中杂交,然后在约50℃、优选约55℃、优选约60℃并且甚至更优选约65℃于1x ssc、0.1%sds中进行一次或多次洗涤。
[0074]
高度严格的条件包括,例如,在约68℃于5x ssc/5x denhardt溶液/1.0%sds中杂交,并且在室温于0.2x ssc/0.1%sds中进行洗涤。可选地,可以在42℃进行洗涤。
[0075]
本领域技术人员将知道哪些条件适合于严格和高度严格的杂交条件。关于这些条件的其他指导可以容易地在本领域中获得,例如在sambrook等人,1989,molecular cloning,a laboratory manual,cold spring harbor press),n.y.;以及ausubel等人(编辑),sambrook and russell(2001)"molecular cloning:a laboratory manual(3rd edition),cold spring harbor laboratory,cold spring harbor laboratory press,new york 1995,current protocols in molecular biology,(john wiley&sons,n.y.)。
[0076]
当然,仅与poly a序列(诸如mrna的3'末端poly(a)段(tract))或者与t(或u)存在物的互补节段(stretch)杂交的多核苷酸不包括在用于特异性地与本发明核酸的一部分杂交的本发明多核苷酸中,因为这种多核苷酸将会与含有poly(a)节段或其互补的任何核酸分子杂交(例如,实际上是任何双链cdna克隆)。
[0077]
同样地,“靶序列”是指要被靶向的核酸内的核苷酸顺序,例如,其中将引入或检测改变。例如,靶序列是dna双链体的第一链所包含的核苷酸的顺序。
[0078]“核酸内切酶”是在结合到其识别位点时水解双链体dna的至少一条链的酶。核酸内切酶在本文中应理解为位点特异性核酸内切酶。限制性核酸内切酶在本文中应理解为同时水解双链体的两条链以在dna中引入双链断裂的核酸内切酶。“切口”核酸内切酶是仅水解双链体的一条链以产生被“切口”而不是切割的dna分子的核酸内切酶。
[0079]
引物结合位点在本文中定义为在双链化时包含可以选择性地与引物序列杂交的引物结合序列的位点。因此,引物结合序列优选地是单链dna序列。
[0080]
核酸内切酶识别位点在本文中定义为包含特定序列,当双链化时,核酸内切酶可以与特定序列结合并随后水解dna的至少一条链。由核酸内切酶识别的特定序列可以位于双链体dna的第一链或第二链中。由核酸内切酶产生的双链或单链断裂可以位于核酸内切酶识别位点内。优选地,断裂可以与核酸内切酶识别序列、或核酸内切酶识别序列上游下游的一个或多个(例如,1、2、3、4、5、6、7、8、9、10、11、12、13、14、15或16)碱基直接相邻。
具体实施方式
[0081]
在第一方面,本发明涉及一种用于生产具有感兴趣的序列的一个或多个单链寡核苷酸的方法,其中所述方法包含以下步骤:
[0082]
a)提供至少一个单链或双链核酸前体,其包含第一链和任选地包含与第一链互补的第二链,其中所述第一链在5'至3'方向上包含以下元件:
[0083]
(1)第一引物结合位点;
[0084]
(2)第一核酸内切酶识别位点;
[0085]
(3)感兴趣的序列;
[0086]
(4)第二核酸内切酶识别位点;以及
[0087]
(5)第二引物结合位点;
[0088]
其中第一核酸内切酶识别位点被设计成使得在双链化之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架;并且,
[0089]
其中第二核酸内切酶识别位点被设计成使得在双链化之后,第二核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架;
[0090]
b)使用能够与第一引物结合位点杂交的第一引物以及能够与第二引物结合位点杂交的第二引物,通过扩增方法来扩增步骤a)的前体;
[0091]
c)用第一核酸内切酶和第二核酸内切酶消化在步骤b)中获得的扩增的双链前体,以通过在感兴趣的序列的紧接上游和下游切割糖-磷酸骨架来生产扩增的双链核酸前体;以及
[0092]
e)使扩增的双链前体变性,从而释放具有感兴趣的序列的单链寡核苷酸。
[0093]
本发明的方法中可以包括额外的步骤,诸如额外的纯化步骤、或所得产物的(长期或短期)储存、或任何其他合适的额外的方法步骤。
[0094]
第一链包含感兴趣的序列。因此,第一链在本文中应理解为核酸前体的链或通过本发明方法步骤b)由此扩增的核酸的链,其包含感兴趣的序列。第二链包含与感兴趣的序列互补的序列。第二链在本文中应理解为核酸前体的链或通过本发明方法步骤b)由此扩增的核酸的链,其与第一链互补。
[0095]
在本文中应理解,第一链的第一引物结合位点包含第一引物结合序列的反向互补,使得互补链(本文中也指示为第二链)将在该第一引物结合位点内包含第一引物结合序列,第一引物可以选择性地与第一引物结合位点退火。在本文中还应理解,第一链的第二引物结合位点在第一链中包含第二引物结合序列,第二引物可以选择性地与第一链退火。优选地,第一引物可以选择性地仅与第一引物结合序列退火,并且第二引物可以选择性地仅与第二引物结合序列退火。任选地,第一引物可以与第一和第二引物结合序列二者退火,并且第二引物可以与第一和第二引物结合序列二者退火,从这个意义上来说,第一和第二引
物可以相同或相似。任选地,(第一和第二)引物可以选择性地仅与第一和第二引物结合位点二者退火。
[0096]
优选地,感兴趣的序列不包含第一和/或第二核酸内切酶识别位点或其反向互补。
[0097]
在优选的实施方案中,本发明的方法进一步包含一个或多个步骤,以便将包含与感兴趣的序列互补的序列的寡核苷酸与第一链、或与第一链的包含感兴趣的序列的其余部分分离。优选地,这是通过在以下各项中添加使第二链、或第二链的至少包含与感兴趣的序列互补的序列的其余部分固定的步骤d)来实现:
[0098]
i)在扩增步骤b)与消化步骤c)之间,
[0099]
ii)在消化步骤c)与变性步骤e)之间;或者,
[0100]
iii)在变性步骤e)之后。
[0101]
优选地,该固定步骤涉及将第二链、或其包含与感兴趣的序列互补的序列的部分亲和捕获在固体支持物上。这可能需要对第二链的整体、或其包含与感兴趣的序列互补的序列的部分进行标签化。可以使用包含亲和标签的、本发明方法步骤b)中的第二引物来实现对第二链的整体进行标签化,以生产包含标签的扩增的双链核酸前体。
[0102]
亲和标签可以至少存在于第二引物上。在本文中应进一步理解,亲和标签可以存在于第一引物和第二引物二者上。可选地,亲和标签不存在于第一引物上,例如,亲和标签仅存在于第二引物上。
[0103]
在另一实施方案中,在扩增之前,步骤b)中使用的第二引物可以存在于如本文所指定的固体支持物上,其中步骤b)中的扩增在固体支持物上进行,从而产生通过第二链附接到固体支持物的扩增子。在该实施方案内,用于扩增的第一引物可以与固体支持物分开提供,例如,可以存在于溶液中,并且第二引物可以例如通过共价键链接到固体支持物、或可以通过如本文中所进一步详述的亲和捕获固定。
[0104]
向本发明方法添加将第二链、或其包含感兴趣的序列的反向互补的部分去除的进一步步骤,以获得具有感兴趣的序列的单链寡核苷酸。优选地,在如上文中所定义的选项i)或ii)中的变性步骤之后、或在如上文中所定义的选项iii)中的固定步骤之后,添加所述去除步骤。优选地,在该实施方案内,设计前体或方法,由此使得对如本发明方法步骤c)中所定义的扩增的双链前体的消化将保持始自标签并包括感兴趣的序列的第二链的糖-磷酸骨架完整。
[0105]
因此,本发明方法的优选实施方案包含以下步骤:
[0106]
a)提供至少一个单链或双链核酸前体,其包含第一链和任选地包含与第一链互补的第二链,其中所述第一链在5'至3'方向上包含以下元件:
[0107]
(1)第一引物结合位点;
[0108]
(2)第一核酸内切酶识别位点;
[0109]
(3)感兴趣的序列;
[0110]
(4)第二核酸内切酶识别位点;以及
[0111]
(5)第二引物结合位点;
[0112]
其中第一引物可以选择性地仅与第一引物结合位点退火,并且第二引物可以选择性地仅与第二引物结合位点退火;
[0113]
其中感兴趣的序列不包含第一和第二核酸内切酶识别位点或其反向互补,
[0114]
其中第一核酸内切酶识别位点被设计成使得在双链化之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架;并且,
[0115]
其中第二核酸内切酶识别位点被设计成使得在双链化之后,第二核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架;
[0116]
b)使用能够与第一引物结合位点杂交的第一引物以及能够与第二引物结合位点杂交的第二引物,通过扩增方法来扩增步骤a)的前体,其中至少第二引物包含亲和标签,以生产包含标签的扩增的双链核酸前体,优选地,亲和标签不存在于第一引物上;
[0117]
c)用所述第一核酸内切酶和所述第二核酸内切酶消化在步骤b)中获得的所述扩增的双链前体,以产生具有所述感兴趣的序列的紧接上游和下游的糖-磷酸骨架被切割、并且具有始自所述标签并包括与所述感兴趣的序列互补的序列的完整糖-磷酸骨架的扩增的双链核酸前体;
[0118]
d)通过亲和捕获标签化的互补第二链将扩增的双链核酸前体固定在固体支持物上;
[0119]
e)使扩增的双链前体变性,从而释放具有感兴趣的序列的单链寡核苷酸;以及
[0120]
f)将固体支持物去除,以获得具有感兴趣的序列的单链寡核苷酸。
[0121]
图1描绘了本发明的优选实施方案的示意图。本领域技术人员理解,本发明的方法可以包含如上文中所详述的步骤。然而,对于本发明而言,并非必须按照上文中指定的顺序进行步骤。在优选的实施方案中,颠倒步骤c)和步骤d)。在可选的实施方案中,颠倒步骤d)和步骤e)。
[0122]
因此,在本发明优选实施方案中,方法可以包含按以下顺序的上文中指定的(以及下文中进一步详述的)步骤:
[0123]
i)步骤a),步骤b),步骤c),步骤d),步骤e)和步骤f);或者
[0124]
ii)步骤a),步骤b),步骤d),步骤c),步骤e)和步骤f);或者
[0125]
iii)步骤a),步骤b),步骤c),步骤e),步骤d)和步骤f)。
[0126]
因此,任选地,本发明的方法可以包含以下后续步骤:
[0127]
a)提供至少一个单链或双链核酸前体,其包含第一链和任选地包含与第一链互补的第二链,其中所述第一链在5'至3'方向上包含以下元件:
[0128]
(1)第一引物结合位点;
[0129]
(2)第一核酸内切酶识别位点;
[0130]
(3)感兴趣的序列;
[0131]
(4)第二核酸内切酶识别位点;以及
[0132]
(5)第二引物结合位点;
[0133]
其中第一引物可以选择性地仅与第一引物结合位点退火,并且第二引物可以选择性地仅与第二引物结合位点退火;
[0134]
其中感兴趣的序列不包含第一和第二核酸内切酶识别位点或其反向互补,
[0135]
其中第一核酸内切酶识别位点被设计成使得在双链化之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架;并且,
[0136]
其中第二核酸内切酶识别位点被设计成使得在双链化之后,第二核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架;
[0137]
b)使用能够与第一引物结合位点杂交的第一引物以及能够与第二引物结合位点杂交的第二引物,通过扩增方法来扩增前体,其中第二引物包含亲和标签,以生产包含标签的扩增的双链核酸前体,其中优选地,亲和标签不存在于第一引物上;
[0138]
d)通过亲和捕获标签化的互补第二链将扩增的双链核酸前体固定在固体支持物上;
[0139]
c)用所述第一核酸内切酶和所述第二核酸内切酶消化所述扩增的双链前体,以产生具有所述感兴趣的序列的紧接上游和下游的糖-磷酸骨架被切割、并且具有始自所述标签并包括与所述感兴趣的序列互补的序列的完整糖-磷酸骨架的扩增的双链核酸前体;
[0140]
e)使扩增的双链前体变性,从而释放具有感兴趣的序列的单链寡核苷酸;以及
[0141]
f)将固体支持物去除,以获得具有感兴趣的序列的单链寡核苷酸。
[0142]
另外,本发明的方法可以包含以下后续步骤:
[0143]
a)提供至少一个单链或双链核酸前体,其包含第一链和任选地包含与第一链互补的第二链,其中所述第一链在5'至3'方向上包含以下元件:
[0144]
(1)第一引物结合位点;
[0145]
(2)第一核酸内切酶识别位点;
[0146]
(3)感兴趣的序列;
[0147]
(4)第二核酸内切酶识别位点;以及
[0148]
(5)第二引物结合位点;
[0149]
其中第一引物可以选择性地仅与第一引物结合位点退火,并且第二引物可以选择性地仅与第二引物结合位点退火;
[0150]
其中感兴趣的序列不包含第一和第二核酸内切酶识别位点或其反向互补,
[0151]
其中第一核酸内切酶识别位点被设计成使得在双链化之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架;并且,
[0152]
其中第二核酸内切酶识别位点被设计成使得在双链化之后,第二核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架;
[0153]
b)使用能够与第一引物结合位点杂交的第一引物以及能够与第二引物结合位点杂交的第二引物,通过扩增方法来扩增前体,其中第二引物包含亲和标签,以生产包含标签的扩增的双链核酸前体,其中优选地,亲和标签不存在于第一引物上;
[0154]
c)用所述第一核酸内切酶和所述第二核酸内切酶消化所述扩增的双链前体,以产生具有所述感兴趣的序列的紧接上游和下游的糖-磷酸骨架被切割、并且具有始自所述标签并包括与所述感兴趣的序列互补的序列的完整糖-磷酸骨架的扩增的双链核酸前体;
[0155]
e)使扩增的双链前体变性,从而释放具有感兴趣的序列的单链寡核苷酸;
[0156]
d)通过亲和捕获将变性扩增双链核酸前体的标签化的互补第二链固定在固体支持物上;以及
[0157]
f)将固体支持物去除,以获得具有感兴趣的序列的单链寡核苷酸。
[0158]
额外的纯化步骤或额外的纯化步骤可以例如包括在步骤a)与步骤b)之间、和/或在步骤b)与步骤c)之间、和/或在步骤c)与步骤d)之间、和/或在步骤d)与步骤e)之间、和/或在步骤e)与步骤f)之间、和/或在步骤d)与步骤c)之间、和/或在步骤e)与步骤d)之间、和/或在步骤b)与步骤d)之间、和/或在步骤c)与步骤e)之间、和/或在步骤d)与步骤f)之
间、和/或在步骤f)之后。
[0159]
可选地,方法可以由以下如上文中所定义的步骤组成:
[0160]
i)步骤a),步骤b),步骤c),步骤d),步骤e)和步骤f);或者
[0161]
ii)步骤a),步骤b),步骤d),步骤c),步骤e)和步骤f);或者
[0162]
iii)步骤a),步骤b),步骤c),步骤e),步骤d)和步骤f)。
[0163]
如果步骤b)中的扩增是在如上文中所详述的固体支持物上进行的,则方法可以包含按以下顺序的上文中指定的(以及下文中进一步详述的)步骤:步骤a),步骤b),步骤c),步骤e)和步骤f)。换句话说,本发明的方法可以包含以下连续的步骤:
[0164]
a)提供至少一个单链或双链核酸前体,其包含第一链和任选地包含与第一链互补的第二链,其中所述第一链在5'至3'方向上包含以下元件:
[0165]
(1)第一引物结合位点;
[0166]
(2)第一核酸内切酶识别位点;
[0167]
(3)感兴趣的序列;
[0168]
(4)第二核酸内切酶识别位点;以及
[0169]
(5)第二引物结合位点;
[0170]
其中第一引物可以选择性地仅与第一引物结合位点退火,并且第二引物可以选择性地仅与第二引物结合位点退火;
[0171]
其中感兴趣的序列不包含第一和第二核酸内切酶识别位点或其反向互补,
[0172]
其中第一核酸内切酶识别位点被设计成使得在双链化之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架;并且,
[0173]
其中第二核酸内切酶识别位点被设计成使得在双链化之后,第二核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架;
[0174]
b)使用能够与第一引物结合位点杂交的第一引物以及能够与第二引物结合位点杂交的第二引物,通过扩增方法来扩增步骤a)的前体,其中将第二引物链接到固体支持物,以生产包含标签的扩增的双链核酸前体;
[0175]
c)用所述第一核酸内切酶和所述第二核酸内切酶消化在步骤b)中获得的所述扩增的双链前体,以产生具有所述感兴趣的序列的紧接上游和下游的糖-磷酸骨架被切割、并且具有始自所述标签并包括与所述感兴趣的序列互补的序列的完整糖-磷酸骨架的扩增的双链核酸前体;
[0176]
e)使扩增的双链前体变性,从而释放具有感兴趣的序列的单链寡核苷酸;以及任选地
[0177]
f)将固体支持物去除,以获得具有感兴趣的序列的单链寡核苷酸。
[0178]
一个或多个额外的纯化步骤可以例如包括在步骤a)与步骤b)之间、和/或在步骤b)与步骤c)之间、和/或在步骤c)与步骤e)之间、和/或在步骤e)与步骤f)之间、和/或在步骤f)之后。可选地,在其中将扩增应用到固体支持物上的该实施方案内,方法在该实施方案中可以由以下如上文中所定义的步骤组成:步骤a),步骤b),步骤c),步骤e)和步骤f)。因为感兴趣的序列已经包含在本发明方法的步骤a)中提供的核酸前体内,所以本发明的方法也可以被认为是一种扩增具有感兴趣的序列的一个或多个单链寡核苷酸的方法。
[0179]
下文对本发明进行更详细的描述:
[0180]
具有感兴趣的序列的寡核苷酸
[0181]
在第一方面,本发明涉及一种用于生产具有感兴趣的序列的一个或多个单链寡核苷酸的方法。单链寡核苷酸在本文中定义为短的单链dna或rna分子。在优选的实施方案中,单链寡核苷酸是单链dna分子。方法特别适合于:使用包含任选地不同序列(例如,不同感兴趣的序列)的多个前体寡核苷酸的初始库(如本文中在“核酸前体”下进一步定义的)作为本发明方法步骤a)中的起始材料,合并生产(即在单一容器中生产)大数目的具有这些任选地不同序列的寡核苷酸。
[0182]
在优选的实施方案中,生产的单链寡核苷酸、或单链寡核苷酸库由以下组成、或各自由以下组成:约20-200个核苷酸,优选约30-180个核苷酸、约40-160个核苷酸、约50-140个核苷酸、约60-120个核苷酸、约70-110个核苷酸、约75-100个核苷酸、约75-95个核苷酸或约80-90个核苷酸。应当理解,这些核苷酸优选是连续的核苷酸。
[0183]
优选地,生产的寡核苷酸、或单链寡核苷酸库由以下组成、或各自由以下组成:至少约20、25、30、35、40、45、50、55、60、65、70、75、80、85、90、95、100、105、110、115、120、125、130、135、140、145、150、155、160、165、170、175、180、185、190、195或200个核苷酸,和/或不具有超过200、195、190、185、180、175、170、165、160、155、150、145、140、135、130、125、120、115、110、105、100、95、90、85、80、75、70、65、60、55、50、45、40、35、30、25或20个核苷酸。
[0184]
在本文中进一步详述的本发明的例示性实施方案中,核酸前体具有选自seq id no:1-seq id no:978的序列,优选地,序列选自seq id no:1-326、seq id no:327-652、和/或seq id no:653-978。最优选地,核酸前体具有选自seq id no:653-978的序列。在该实施方案中,用作起始材料的核酸前体库包含由seq id no:1-seq id no:978表示的这些978个核酸前体的库或由seq id no:1-seq id no:978表示的这些978个核酸前体的库组成。
[0185]
要生产的单链寡核苷酸、或单链寡核苷酸库可以包含感兴趣的序列或由感兴趣的序列组成、或各自可以包含感兴趣的序列或各自由感兴趣的序列组成。优选地,通过本发明的方法生产的单链寡核苷酸、或单链寡核苷酸库由感兴趣的序列组成、或各自由感兴趣的序列组成。特别优选的感兴趣的序列是可以例如作为扩增的引物、作为用于连接、杂交或(在溶液中)捕获的探针、或作为衔接子(adaptor)、或作为体外转录的模板使用的序列。
[0186]
用作引物的感兴趣的序列、或引物寡核苷酸可以包含至少部分地与要扩增的预定靶序列(诸如预定(基因组)dna序列、cdna序列、rna序列、和/或无细胞dna序列)互补的序列。此种序列在本文中被称作互补靶序列。优选地,所述互补靶序列与预定靶序列至少80%、85%、90%、98%或99%互补。最优选地,互补靶序列与预定靶序列完全互补(100%)。优选地,此种互补靶序列是长度为约18、19、20、21、22、23个核苷酸的节段。任选地,用作引物的感兴趣的序列包含其他功能性元件,诸如用于随后的(一个或多个)扩增和/或测序步骤的一个或多个引物结合位点、和/或例如用于样本追踪或分子索引的一个或多个条形码序列(任选地,诸如wo2016/201142中所描述的间断的条形码)、和/或一个或多个简并核苷酸。引物可以是加尾引物,其在本文中应理解为是指在3'端处包含互补靶序列以及尾的引物,所述尾包含一个或多个功能性元件,优选如上文中所指示的功能性元件。可选地,引物可以是诸如us2008/0305478 a1、us2010/0227320 a1、us2016/0068903 a1中所描述的ω引物。此种ω引物通常在引物的3'和5'端二者处包含两个互补靶序列(通常分别是长度为6-60个核苷酸的节段和长度为10-100个核苷酸的节段),两个互补靶序列由环(通常是长度为
12-50个核苷酸的节段)隔开,环不与靶结合并且随后可用作单重pcr的引发部分(section)。
[0187]
本发明的方法特别适合于生产引物寡核苷酸的确定的库,其可以用于例如基于多重寡核苷酸的扩增,诸如多重pcr。此种引物库可以包含引物对或由引物对组成,所述引物对适合于一起用于扩增特定的靶序列。任选地,所述对中的两个引物都是靶特异性的,这在本文中应理解为,引物的至少一部分包含与要扩增的特异序列(其可以是某个基因或其部分)互补的序列。可选地,所述对中的一个引物是所谓的普通引物,其可以能够与对特定靶序列(例如,衔接子中的预定序列)不是特异性的序列退火,而所述对中的另一个引物是靶特异性的。任选地,所述对中的两个引物都是普通引物。如果所述对中的引物是加尾引物,则尾可以包含通用序列,以用于随后用一对普通引物进行的尾pcr。
[0188]
生产的寡核苷酸适合于用作至少10-、20、40-、60-、80-、100-、120-、140-、160-、180-、200-、220-、240-、260-、280-、300-、320-、326-、340-、360、380-、400-、420-、440-、460-、480-、500-、600-、700-、800-、900-、1,000-、2,000-、3,000-、4,000-、5,000-、6,000-、7,000-、8,000-、9,000-、10,000-、20,000-、30,000-、40,000-、50,000-、60,000-、70,000、80,000-、90,000-、100,000-、200,000-、300,000-、400,000-、或500,000-重pcr测定中的引物。n-重pcr测定在本文中应理解为在单个反应容器中的pcr反应,其导致n个不同靶序列的扩增。通过本发明方法生产的引物还可以用于通过合成测序或用于克隆。
[0189]
在本发明方法中生产的寡核苷酸还特别适合于用作探针。因此,感兴趣的序列可以包含探针序列或由探针组成。探针或探针寡核苷酸在本文中应理解为(单独或与一种或多种其他探针组合)用于检测与探针中的序列互补的核苷酸序列(即,靶序列)的存在的寡核苷酸。因此,此类探针序列包含如上文中所定义的互补靶序列,并且可以进一步包含一个或多个引物结合位点和/或一个或多个条形码序列。探针可以进一步包含标签(标记),例如,亲和配体、或放射性或荧光标签。通过本发明方法生产的寡核苷酸探针特别尤其适合用于核酸检测领域,诸如通过杂交或(在溶液中)捕获核酸(杂交捕获探针)的(高通量)核酸检测、靶向变异检测、以及靶向和/或可编程基因组编辑。本发明的方法特别适合于生产确定的探针寡核苷酸库,所述确定的探针寡核苷酸库可以用于例如多重ola。
[0190]
探针可以是ola探针,ola探针与另一探针一起可以用于例如snp或插入缺失检测。如在例如wo2007/100243中所描述的,用于第一和第二探针的杂交的两个靶序列彼此相邻定位,使得探针可以在结合时直接连接,或者这些两个靶序列不相邻但在它们之间留有空位,从而需要空位填充(akhunov et al.theor.appl.genet.2009aug;119(3):507-517)或空位连接(使用如在例如wo00/77260中所描述的合适的第三寡核苷酸)。另外,通过本发明的方法生产的探针也可以是锁式探针(padlock probe)(例如,如在nilsson等人,science.1994sep 30;265(5181):2085-2088中所描述的)、分子倒位(inversion)探针(例如,如在hardenbol等人,nat biotechnol.2003jun;21(6):673-678中所描述的)、或连接器倒位(connector inversion)探针(例如,诸如在akhras等人,plos one.2007;2(9):e915中所描述的),这些探针全部都是通常包含在与靶互补的两个区段(每个的长度通常为约20个核苷酸)的单链核酸分子,并且这些部分通过接头(linker)(例如长度为40个核苷酸的接头)连接。核酸分子与靶序列杂交并连接时(任选地,在空位填充后)被环化。接头序列中存在功能性,可以允许进行扩增和随后的检测。
[0191]
使用如本文中所定义的一个或多个引物的待扩增的特别优选的预定靶序列和/或使用如本文中所定义的一个或多个探针的待检测的特别优选的预定靶序列是具有遗传变异的基因组序列,例如,含有多态性(即,多态性位点)、表示多态性或与多态性相关联的核苷酸序列。术语多态性在本文中是指在群体中出现两个或更多个遗传确定的可选序列或等位基因。在探针的情况下,互补靶序列优选地(至少部分地)与多态性位点的这些两个或更多个遗传确定的可选序列中的仅一个互补。在引物的情况下,互补靶序列优选地(至少部分地)与遗传确定的(例如,在上游或在下游)侧接有此种多态性位点的序列互补。
[0192]
多态性位点可以小到一个碱基对,诸如snp。多态性包括:限制性片段长度多态性、可变数目的串联重复序列(vntr)、高变区、小卫星、二核苷酸重复序列、三核苷酸重复序列、四核苷酸重复序列、简单序列重复序列、以及插入元件(诸如alu)。在探针的情况下,互补靶序列(至少部分地)与两个或更多个遗传确定的可选snp等位基因序列中的仅一个互补。更优选地,在连接探针的情况下,互补靶序列的5'或3'端处的核苷酸与可选(snp)等位基因中的仅一个互补。
[0193]
在优选的实施方案中,生产的寡核苷酸适用于ola测定。本发明的方法导致高质量单链寡核苷酸的生产。此类寡核苷酸特别可用于多重测定,诸如但不限于基于多重寡核苷酸的扩增(诸如多重pcr)、多重捕获杂交、mlpa和多重ola测定。优选地,生产的寡核苷酸适用于例如以下所描述的ola多重测定:us 4,988,617;nilsson et al.(同上);us 5,876,924、wo98/04745;wo98/04746;us6,221,603;us5,521,065;us5,962,223;ep185494bi;us6,027,889;us4,988,617;ep246864b1;us6,156,178;ep745140b1;ep964704b1;wo03/054511;us2003/0119004;us2003/190646;ep1313880;us2003/0032016;ep912761;ep956359;us2003/108913;ep1255871;ep1194770;ep1252334;w096/15271;w097/45559;us2003/0119004a1;us5,470,705;wo 2004/111271;wo2005/021794;wo2005/118847;wo03/052142;van eijk mj(同上);wo2007/100243;wo01/57269;wo03/006677;wo01/061033;wo2004/076692;wo2006/076017;wo2012/019187;wo2012/021749;wo2013/106807;wo2015/154028;wo2015/014962和wo2013/009175。
[0194]
在进一步优选的实施方案中,生产的寡核苷酸适合于用作至少10-、20-、40-、60-、80-、100-、120-、140-、160-、180-、200-、220-、240-、260-、280、300-、320-、326-、340-、360-、380-、400-、420-、440-、460-、480-、500-、600-、700-、800-、900-、1,000、2,000-、3,000-、4,000-、5,000-、6,000-、7,000-、8,000-、9,000-、10,000-、20,000-、30,000-、40,000-、50,000-、60,000-、70,000-、80,000-、90,000-、100,000-、200,000-、300,000-、400,000-或500,000-重ola测定中的引物。优选地,生产的寡核苷酸适用于至少300-重ola测定、并且甚至更优选至少326-重ola测定。
[0195]
通过本发明方法生产的寡核苷酸还可以用作单链衔接子或用于制备部分或完全双链衔接子(诸如但不限于y形衔接子)。部分或完全双链衔接子可以通过使两个部分或完全互补的单链寡核苷酸退火来形成。优选地,用作衔接子的寡核苷酸包含功能性元件,诸如但不限于:用于(一个或多个)扩增步骤和/或测序的一个或多个引物结合位点、和/或例如用于样本追踪或分子索引的一个或多个条形码序列(任选地,诸如wo2016/201142中所描述的间断的条形码)、和/或一个或多个简并核苷酸。
[0196]
核酸前体
[0197]
本发明方法的第一步骤是提供至少一个单链或双链核酸前体,其包含第一链和任选地包含与第一链互补的第二链。优选地,核酸前体是dna分子。
[0198]
因此,用于本发明方法的核酸前体可以是包含第一链的单链核酸前体。可选地,用于本发明的核酸前体可以是包含第一链和与第一链互补的第二链的双链核酸前体。优选地,核酸前体的任选的第二链与第一链至少80%、85%、90%、98%或99%互补。最优选地,任选的第二链在其整个长度上与第一链完全互补(100%)。
[0199]
优选地,核酸前体是单链核酸前体,并且最优选地,核酸前体是单链dna核酸前体。
[0200]
核酸前体的长度为至少约50、60、70、80或90个核苷酸,优选地,长度为至多约500、450、400、350或300个核苷酸,诸如在50与500、50与400、50与350、50与300、80与500、80与400、80与350、80与300个核苷酸之间。
[0201]
优选地,第一链在5'至3'方向上包含以下五个元件或由以下五个元件组成:
[0202]
(1)第一引物结合位点;
[0203]
(2)第一核酸内切酶识别位点;
[0204]
(3)感兴趣的序列;
[0205]
(4)第二核酸内切酶识别位点;以及
[0206]
(5)第二引物结合位点。
[0207]
这些五个元件可以是五个不同的元件(如图2b所例示),或者一个或多个可以部分或完全重叠的元件(图2a)。例如,第一核酸内切酶识别位点可以部分或完全地包含在第一引物结合序列的反向互补序列内,和/或第二核酸内切酶识别位点可以部分或完全地包含在第二引物结合序列内。因此,相同的序列可以充当引物结合序列以及核酸内切酶识别位点(图2a)。
[0208]
因此,第一链包含第一引物结合位点(具有第一引物结合序列的反向互补序列;在双链化时,互补链将包含第一引物可以与其退火的第一引物结合序列)以及第二引物结合位点(具有第二引物可以与其退火的第二引物结合序列)。在第一链双链化(以获得第一链和互补的第二链)时,第一引物可以选择性地仅与第一引物结合位点退火(例如,杂交),并且第二引物可以选择性地仅与第二引物结合位点退火(例如,杂交)。换句话说,第一引物将不会与核酸前体和/或其互补退火,除了第一引物结合位点之外。类似地,第二引物将不会与核酸前体或其互补退火,除了第二引物结合位点之外。任选地,第一和第二引物与第一和第二引物结合位点二者退火,从这个意义上来说,第一和第二引物可以相同或相似。另外,第一和第二引物结合位点的序列可以相同。换句话说,第一引物结合序列可以与第二引物结合序列相同。
[0209]
核酸前体包含如上文中所定义的感兴趣的序列。在进一步优选的实施方案中,提供了两个或更多个核酸前体的库。优选地,所述库包含至少2、3、4、5、10、50、100、150、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、978、1000、1050、1100、1150、1200、1300、1400、1500、2000、3000、4,000、5,000、6,000、7,000、8,000、9,000、10,000、20,000、30,000、40,000、50,000、60,000、70,000、80,000、90,000、100,000、200,000、300,000、400,000、500,000、600,000、700、000、800,000、900,000、1,000,000、1,100,000、1,200,000、1,300,000、1,400,000、或1,500,000个核酸前体。
[0210]
该核酸前体库的核酸序列在所述库的全部或部分核酸前体之间可以不同。这些核
酸前体在感兴趣的序列的核苷酸序列中、在(一个或多个)引物结合位点和/或(一个或多个)核酸内切酶识别位点中可以不同。核酸前体库可以包含至少2、3、4、5、10、50、100、150、200、250、300、350、400、450、500、550、600、650、700、750、800、850、900、950、978、1000、1050、1100、1150、1200、1300、1400、1500或2000、3000、4,000、5,000、6,000、7,000、8,000、9,000、10,000、20,000、30,000、40,000、50,000、60,000、70,000、80,000、90,000、100,000、200,000、300,000、400,000、500,000、600,000、700、000、800,000、900,000、1,000,000、1,100,000、1,200,000、1,300,000、1,400,000、或1,500,000个独特序列。包含至少2个独特序列的核酸前体库在本文中应理解为包含至少2个核酸前体的库,所述至少2个核酸前体在它们的整个长度上不具有相同的核苷酸序列,即,它们的核苷酸序列在至少一个核苷酸位置上不同。
[0211]
在优选的实施方案中,核酸前体的初始库可以含有约2%、10%、15%、20%、25%、30%、40%、50%、60%、70%、75%、80%、90%、95%、98%或100%的独特序列。核酸前体初始库在本文中应理解为扩增步骤之前的核酸前体库。更优选地,核酸前体初始库可以含有约75%或100%的独特序列,在此含有约75%独特序列的库是最优选的。此种库通常是生产用于(多重)ola测定的探针的库,其中优选地,对于每个snp 2,使用不同的等位基因探针和一个基因座探针,并且其中这些探针以以下比率存在于连接测定中:第一等位基因探针1:第二等位基因探针2:基因座探针为1:1:2,以便产生等摩尔量的等位基因探针和基因座探针。因此,在一个优选的实施方案中,核酸前体初始库可以含有比率为约1:1:2的独特序列。可选地,核酸前体初始库可以含有比率约为1:1的独特序列(用于寡核苷酸生产,以用于仅使用邻接的、相邻的或更远距离隔开的基因座特异性探针的基于多重寡核苷酸的扩增或ola测定)。
[0212]
优选地,核酸前体的独特序列选自seq id no:1-seq id no:978。另外,至少一个序列可以选自seq id no:1-326,一个序列可以选自seq id no:327-652,和/或一个序列可以选自seq id no:653-978。
[0213]
对于库内的寡核苷酸前体中的每一个,核酸前体中的每一个的第一引物结合位点序列可以相同。另外或可选地,对于库内的核酸前体中的每一个,库中寡核苷酸前体中的每一个的第二引物结合位点的序列可以相同。另外或可选地,对于库内的核酸前体中的每一个,库中寡核苷酸前体中的每一个的第一核酸内切酶识别位点可以相同。另外或可选地,对于库内的核酸前体中的每一个,库中寡核苷酸前体中的每一个的第二核酸内切酶识别位点可以相同。如本文中较早所指示的,在任选的实施方案中,第一和第二引物以及引物结合位点可以相同或高度相似,使得第一引物还可以与第二引物结合位点退火并且反之亦然,以允许对核酸前体进行扩增。在任选的实施方案中,其中用于本发明方法的第一和第二核酸内切酶是限制性酶,尽管第一和第二核酸内切酶识别位点彼此呈反向互补方向,但是它们可以相同。换句话说,在该实施方案中,第一链中的第一核酸内切酶识别位点的核苷酸序列是第一链中的第二核酸内切酶识别位点的核苷酸序列的反向互补。
[0214]
任选地,按照以下方式来设计库中的核酸前体:允许根据一个或多个特定引物对的选择来生产寡核苷酸的特异性亚组。举例来说,库内核酸前体的特定亚组可以包含特定的引物结合位点组合。优选地,这些引物结合位点组合包含一个或多个引物结合序列,所述一个或多个引物结合序列在这些引物结合序列的5'端处至少以2、3、4、5、6或更多个核苷酸
发生变化(本在文中被称为核苷酸的可变部分),从而允许使用在其3'端处具有相应(watson-crick)的1、2、3、4、5、6或更多个核苷酸的引物进行特异性亚组的扩增。
[0215]
例如,核酸前体的两个不同亚组的第一和/或第二引物结合位点包含通用部分(两个亚组的核苷酸序列相等)和可变部分(两个亚组的核苷酸序列不同)。优选地,该通用部分具有至少18个核苷酸,并且该可变部分的长度为1、2、3、4或更多个核苷酸。可变部分位于引物结合序列的5'末端部分处,并且通用部分位于引物结合序列的3'末端部分处(两个例示性实施方案参见图3和图4)。在扩增此类核酸前体时,可以使用在其3'端(与引物结合序列的可变部分互补并能够与其退火)具有选择性核苷酸的一个或多个引物。此类选择性核苷酸的存在或缺乏将确定将对前体的哪个亚组或任选地所有亚组进行扩增。举例来说,使用没有选择性核苷酸(+0/+0)的引物,即,仅包含与引物结合序列的18个核苷酸长的通用部分互补的序列的引物,将允许对两个亚组一起进行扩增。使用包含例如在两个引物对的3'端处的两个选择性核苷酸(+2/+2)、或在同与引物结合序列的通用部分互补的18个核苷酸长的核苷酸相邻的一对引物中的一个上的两个选择性核苷酸(+0/+2或+2/+0),将允许对亚组中的任何一个进行扩增。因此,在特定实例中,引物的两个选择性核苷酸与可变部分的两个核苷酸互补,位于与引物结合位点的通用部分的18个核苷酸直接相邻。
[0216]
因此,仅分别与第一和第二引物结合序列的通用部分退火的引物对允许对所有亚组进行扩增,即,对核酸前体的完全初始库进行扩增。
[0217]
相比之下,包含(部分或完全地)与引物结合序列的可变部分退火的、并且任选地还(部分或完全地)与引物结合序列的通用部分退火的至少一个引物的引物对允许对一个或多个亚组进行扩增。在本文中应当理解,该引物对的第二引物可以仅与另一引物结合序列的通用部分退火或可以(部分或完全地)与另一引物结合序列的可变部分退火,并且任选地还(部分或完全地)与另一引物结合序列的通用部分退火。
[0218]
在优选的实施方案中,引物结合序列的通用部分包含至少16、17、18、19、20、21、22、23或至少24个核苷酸。另外,引物结合序列的可变部分包含至少0、1、2、3、4、5、6、7、8、9或至少10个核苷酸。
[0219]
另外或可选地,核酸前体可以包含具有如本文中所详述的可变部分和通用部分的引物结合位点,其中引物可以例如仅与可变部分结合以允许进行扩增。在该实施方案中,可变部分可以优选地包含至少16、17、18、19、20、21、22、23或至少24个核苷酸。足以使引物有效退火的此种相对长的可变部分也可以被认为是其自身的单独的引物结合位点。换句话说,库中的核酸前体因此可以包含(紧邻第一和第二引物结合位点)一个或两个额外的引物结合位点(例示性实施方案参见图5和图6)。更具体地,库中的核酸前体(的第一链)可以在第一引物结合序列的反向互补的上游或5'端处包含第三引物结合序列的反向互补,和/或可以在第二引物结合序列的下游或3'端处包含第四引物结合序列。库内的核酸前体可以被设计成使得特定亚组包含特定的第一和第二引物结合位点组合,而包括该特定亚组的更大亚组包含特定的第三和第四引物结合位点组合。在本文中应进一步理解,第一、第二、第三和第四引物结合位点中的至少一个可以再次包含如本文中所详述的可变部分和通用部分,从而允许通过可变部分的修饰以及特定引物对的使用来对特异性亚组进行扩增。
[0220]
另外,前体的第一链内的引物结合位点的可变部分可以在第一核酸内切酶识别位点的下游和/或第二核酸内切酶识别位点的上游(如图3和图5所例示),使得第一核酸内切
酶在第一引物结合位点的可变部分下游切割第一链的糖-磷酸骨架,和/或第二核酸内切酶在第二引物结合位点的可变部分上游切割第一链的dna。
[0221]
用于本发明方法的核酸前体进一步包含第一核酸内切酶识别位点和第二核酸内切酶识别位点。
[0222]
核酸前体包含第一核酸内切酶识别位点,所述第一核酸内切酶识别位点被设计成使得在双链化之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架。措辞“在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架”意味着在感兴趣的序列的5'-核苷酸与所述5'-核苷酸的上游(或5'侧)的第一核苷酸之间切割糖-磷酸骨架。结果是,感兴趣的序列的5'-末端核苷酸以及所述5'-核苷酸的下游(或3'侧)的序列不再是包含第一引物结合位点的反向互补以及第一核酸内切酶识别位点的dna链的部分。
[0223]
核酸前体包含第二核酸内切酶识别位点,所述第二核酸内切酶识别位点被设计成使得在双链化之后,第二核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架。措辞“在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架”意味着在感兴趣的序列的3'-核苷酸与所述3'-核苷酸的下游(或3'侧)的第一核苷酸之间切割糖-磷酸骨架。结果是,感兴趣的序列的3'-核苷酸以及所述3'-核苷酸的上游的序列不再是包含第二引物结合位点以及第二核酸内切酶识别位点的dna链的部分。因此,识别双链化前体的第一核酸内切酶识别位点的第一核酸内切酶在感兴趣的序列的紧接上游切割dna。类似地,识别双链化前体的第二核酸内切酶识别位点的第二核酸内切酶在感兴趣的序列紧接下游切割dna。
[0224]
如本文中所详述的,核酸内切酶在感兴趣的序列的直接上游(第一核酸内切酶)或直接下游(第二核酸内切酶)切割第一链的糖-磷酸骨架。这可以通过使用本领域已知的所谓“外部切割器(outside cutter)”来实现。此类外部切割器可以分别在核酸内切酶识别位点内的第一和/或第二核酸内切酶识别序列直接相邻的第一链切割糖-磷酸骨架。可选地,外部切割器可以在所述酶的识别序列之外至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15或16个核苷酸切割糖-磷酸骨架。举例来说,在第一核酸内切酶切割了核酸内切酶识别序列之外的10个核苷酸的情况下,将有10个核苷酸存在于核酸内切酶识别序列与感兴趣的序列之间。如本文中所指示的,位于核酸内切酶识别序列与感兴趣的序列之间的这些核苷酸可以是第一和/或第二引物结合位点的一部分,任选地,可以构成第一和/或第二引物结合位点的可变部分。第一核酸内切酶和/或第二核酸内切酶可以是切口核酸内切酶或限制性核酸内切酶。优选地,感兴趣的序列被设计成使得、并且用于本发明方法的核酸内切酶被选择成使得,在本发明方法的消化步骤后,感兴趣的序列保持完整。
[0225]
在将第二链或其至少包含感兴趣的序列的反向互补的其余部分与第一链或其至少包括感兴趣的序列的其余部分分离的情况下,本发明的方法包含对扩增的双链前体的第二链进行标签化。如本文中所进一步详述的,该标签优选地位于扩增的双链前体的第二链的5'端处,并且可以在扩增步骤中通过使用标记的引物来引入。在该实施方案内,优选地,设计前体或方法,由此使得对本发明方法的扩增的双链前体消化后,第二链始自标签并包括感兴趣的序列的反向互补的糖-磷酸骨架保持完整。另外,可以在第二链的与感兴趣的序列互补的序列的3
’
切割糖-磷酸骨架。因此,优选的是,不切割与感兴趣的序列互补的序列。然而,在本发明内考虑到,可以在与感兴趣的序列互补的序列的3'端附近切割其糖-磷酸骨架,例如,可以在与感兴趣的序列互补的序列的3'端处的最后1、2、3、4、5、6、7、8、9或10个核
苷酸之前切割糖磷酸骨架。
[0226]
允许第二链始自标签并包括感兴趣的序列的反向互补的糖-磷酸骨架保持完整的一种可能的前体的设计是,选择被设计成要被切口核酸内切酶识别的第二限制性识别位点,在这种情况下切口核酸内切酶被定向成仅在感兴趣的序列的紧接下游切口第一链。然后,所述切口核酸内切酶用作本发明方法的消化步骤中的第二核酸内切酶。
[0227]
举例来说,在第一核酸内切酶是nt.alwi(new england biolabs)(其能够催化其识别序列ggatc之外4个碱基的单链断裂(即,5'
…
ggatcnnnnn...3'))的情况下,第一核酸内切酶识别位点(在5'至3'方向上)包含ggatcnnnn或由ggatcnnnn组成,其与感兴趣的序列的5'-核苷酸紧接相邻。举例来说,在第二核酸内切酶是nb.bsrdi(new england biolabs)(其催化与cattgc的5'端直接相邻的单链断裂(即,5'
…
nncattgc
…
3'))的情况下,第二re识别位点(在5'至3'方向上)包含cattgc或由cattgc组成,并且与感兴趣的序列的3'-核苷酸紧接相邻。
[0228]
允许第二链始自标签并包括感兴趣的序列的反向互补的糖-磷酸骨架保持完整的一种可能的方法的设计是,选择具有不能被核酸内切酶切割的化学性质的第二引物。此种化学性质是本领域已知的,并且可以选自但不限于基于硫代磷酸酯(ps)键、甲基化(例如,n6-甲基腺苷或ma、5-甲基胞嘧啶或mc、5-羟甲基胞嘧啶或hmc)和锁核酸(lna)的化学性质。在该特定实施方案中,第二核酸内切酶可以是限制性核酸内切酶,其能够在感兴趣的序列的3'-端核苷酸与第二核酸内切酶识别位点的5'-端核苷酸之间切割第一链、以及在感兴趣的序列的反向互补的5'-端核苷酸与第二核酸内切酶识别位点的3
’-
端核苷酸或该位置在第二链上的5'的任何位置之间切割第二链。第二引物应被设计成使得所生产的扩增子的第二链对通过所选的第二(限制性)核酸内切酶进行的切割是惰性的。这可以通过使用修饰过的第二引物来设想,所述修饰过的第二引物在第二链上的第二(限制性)核酸内切酶正常切割的位置处产生具有耐核酸内切酶化学性质的扩增子。
[0229]
扩增
[0230]
本发明的方法包含一种使用第一引物和第二引物通过扩增方法来扩增如本文中所定义的核酸前体的步骤。核酸前体的扩增优选地导致核酸前体的丰度增加至少100倍,优选至少500、1000或甚至至少5000倍。本发明方法中的扩增步骤导致生成(扩增的)双链核酸前体。
[0231]
任何扩增方法都可以适用于本发明的方法,诸如聚合酶链反应以及等温扩增方法。在使用pcr来扩增核酸前体的情况下,优选地使用高保真dna聚合酶来减少pcr过程中错误掺入的数目。
[0232]
优选地,扩增方法是等温扩增方法。若干等温扩增方法是本领域已知的,诸如环介导的等温扩增(lamp)、链置换扩增(sda)、切口酶扩增反应(near)、解旋酶依赖性扩增(hda)和重组酶聚合酶扩增(rpa),并且本文中描述的本发明不限于单一等温扩增方法。优选的等温扩增方法是重组酶聚合酶扩增(rpa)或解旋酶依赖性扩增(hda)。
[0233]
解旋酶依赖性扩增利用解旋酶的双链dna解链活性来分离链,使得能够通过链置换dna聚合酶进行引物退火和延伸。hda是本领域已知的。例如,hda方法可以包含如在us9074248中所描述的以下步骤:
[0234]-将合适的缓冲剂、核酸前体;第一和第二引物;解旋酶;以及三磷酸脱氧核苷酸
(dntp)组合;
[0235]-将反应混合物在优选低于引物的熔融温度约5摄氏度至高于引物的熔融温度约3摄氏度之间的温度温育;以及
[0236]-得到扩增的模板核酸。
[0237]
特别优选的扩增方法是重组酶聚合酶扩增(rpa)。rpa是本领域已知的,并且例如可以如在piepenburg等人(2008)、wo2003/072805、wo2005/118853、wo2007/096702、wo2008/035205、wo2010/135310、wo2010/141940、wo2011/038197、wo2012/138989中所描述的、和/或根据制造条件使用twistdx的twistamp basic试剂盒来进行。
[0238]
简而言之,在适合于rpa发生的缓冲剂中,使如本文中所定义的核酸前体与第一和第二引物以及至少三种酶(即,至少重组酶、聚合酶和单链dna结合蛋白(ssb))接触。优选地,在添加酶之前,使(一种或多种)核酸前体与第一和第二引物接触。在下文中详细概述了pra的实例。然而,本发明决不限于下文中详述的rpa反应,并且本领域技术人员理解该方案的变化是在本发明的范围内。
[0239]
例如,将2.4μl的第一引物(10μm)、2.4μl的第二引物(10μm)和0.01-0.05pmol的核酸前体在h2o中混合至总体积为18μl。随后可以添加缓冲剂,特别如果用于rpa的酶处于冷冻干燥状态,则可以将例如29.5μl的再水化缓冲剂(rehydration buffer)添加到上述18μl的总体积中,该缓冲剂具有以下成分:
[0240]
0-60mm tris,例如,25mm tris
[0241]
50-150mm醋酸钾,例如,100mm醋酸钾
[0242]
0.3-7.5w/v聚乙二醇,例如,5.46%w/v peg 35kda。
[0243]
任选地,将再水化溶液(包含缓冲剂、引物和(一种或多种)核酸前体)涡旋并短暂离心。随后,可以将总体积为47.5μl的再水化溶液转移到碱性rpa冷冻干燥的反应沉淀物中,所述反应沉淀物优选地包含以下组分(其中,所指示浓度如冷冻干燥前或如重构后所示):
[0244]-至少一种重组酶(例如,100-350ng/μl uvsx重组酶,诸如260ng/μl,优选噬菌体t6 uvsx重组酶);
[0245]-至少一种单链dna结合蛋白(150-800ng/μl gp32,诸如254ng/μl,优选噬菌体rb69 gp32);
[0246]-至少一种dna聚合酶(例如,30-150ng/μl枯草芽孢杆菌(bacillus subtilis)pol i(bsu)聚合酶或金黄色葡萄球菌(s.aureus)pol i大片段(sau聚合酶),诸如90ng/μl);
[0247]-dntp或dntp与ddntp的混合物(150-400μm dntp,诸如240μm);
[0248]-拥挤剂(crowding agent)(例如,聚乙二醇,优选1.5-5%w/v peg 35kda,诸如2.28%w/v peg 35kda,任选地与2.5%-7.5%重量/体积的海藻糖(诸如5.7%w/v海藻糖)组合);
[0249]-缓冲剂(例如,0-60mm tris缓冲剂,诸如25mm tris);
[0250]-还原剂(例如,1-10mm dtt,诸如5mm dtt);
[0251]-atp或atp类似物(例如,1.5-3.5mm atp,诸如2.5mm atp);
[0252]-任选地至少一种重组酶加载蛋白(例如,50-200ng/μl uvsy,优选噬菌体rb69 uvsy,诸如88ng噬菌体rb69 uvsy);
[0253]-磷酸肌酸(例如,20-75mm,诸如50mm磷酸肌酸);以及
[0254]-肌酸激酶(例如,10-200ng/μl,诸如100ng/μl)。
[0255]
反应混合物可以进一步包含50-200ng/μl的核酸外切酶iii(exoiii)、核酸内切酶iv(nfo)或8-氧代鸟嘌呤dna糖基化酶(8-oxoguanine dna glycosylase)(fpg)。
[0256]
可以将镁添加到反应混合物中以开始rpa反应,例如,可以添加醋酸镁至反应混合物中的终浓度为8-16mm(例如,可以将2μl 280mm醋酸镁添加到上文中例示的47.5μl的反应体积中)。任选地,醋酸镁已经存在于反应混合物中,即,随后不添加,但是例如同上文中定义的再水化溶液的其他成分一起与(一种或多种)核酸前体接触。将反应温育直到实现期望的扩增程度。在使寡核苷酸前体与如上文中所指示的这些酶、引物和缓冲剂组分接触后,优选地将混合物在约37℃(优选在25℃至42℃之间)温育约1小时。优选地,rpa导致核酸前体扩增至少100倍,优选至少200、300或甚至至少400倍,例如,约500倍。
[0257]
用于rpa的其他方案可以同样适合于核酸前体的扩增。更具体地,可以使用其他重组酶,诸如但不限于大肠杆菌(e.coli)reca、或来自任何门(例如,rad51)的任何同源蛋白或蛋白复合物、或rect、或reco、或uvx,诸如aeh1 uvx、t4 uvsx、t6 uvsx和rb69 uvx。聚合酶可以是真核聚合酶或原核聚合酶。原核聚合酶至少包括大肠杆菌pol i、大肠杆菌pol ii、大肠杆菌pol iii、大肠杆菌pol iv和大肠杆菌polv。真核聚合酶包括例如选自以下的多蛋白聚合酶复合物:pol-、pol-β、pol-δ和pol-ε。合适的聚合酶可以是大肠杆菌polv或其他物种的同源物聚合酶。进一步合适的单链dna结合蛋白(ssb)可以是大肠杆菌gp32或aeh1 gp32、t4 gp32、rb69 gp32。要使用的合适的酶浓度为:20μm重组酶、约1-10μm ssb和约1-2μm聚合酶。其他任选的拥挤剂(除了聚乙二醇和/或海藻糖之外)是但不限于聚氧化乙烯、聚乙烯醇、聚苯乙烯、ficoll、右旋糖酐、pvp和白蛋白。在优选的实施方案中,拥挤剂的分子量小于200,000道尔顿。进一步地,拥挤剂可以约0.5%至约15%重量与体积比(w/v)的量存在。
[0258]
用于扩增核酸前体的引物与核酸前体退火至允许引物延伸以使用例如rpa或pcr扩增的程度。具体而言,第一引物(仅)与第一引物结合序列退火,并且第二引物(仅)与第二引物结合序列退火。
[0259]
在优选的实施方案中,第一引物与第一引物结合序列完全互补,并且第二引物与第二引物结合序列完全互补。在引物结合位点具有如本文中所定义的可变部分的情况下,引物可以仅与引物结合序列的通用部分以及任选地与引物结合序列的可变部分的一部分完全互补。可选地,引物可以仅与引物结合序列的可变部分以及任选地与引物结合序列的通用部分的一部分完全互补。类似地,引物可以与引物结合序列的可变部分部分互补,并且与引物结合序列的通用部分部分互补。
[0260]
另外,第一和/或第二引物可以进一步包含额外序列,所述额外序列存在于与引物结合序列互补的序列的5
’
。优选地,所述额外序列可以由互补序列5
′
的1、2、3、4、5、6、7、8、9、10或15个额外核苷酸组成。如本文上文中所指示的,第一链在本文中应理解为包含核酸前体的、或本发明方法步骤b)中获得的扩增子的感兴趣的序列的链。同样地,第二链在本文中应理解为核酸前体的或本发明方法步骤b)中获得的扩增子的链,第二链与第一链互补。如本领域技术人员所理解的,在如本文中所指示的第一引物和第二引物在它们的5'端处包含额外的核苷酸的情况下,本发明方法步骤b)中获得的扩增子的链将长于相应的核酸前体
的链。
[0261]
第一引物和/或第二引物的长度优选为约12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29或30个核苷酸。第一引物和第二引物的长度可以相同或不同。在优选的实施方案中,第一引物的长度优选为约12、13、14、15、16、17、18、19、20、21、22、23、24、25,26、27、28、29或30个核苷酸,并且第二引物的长度优选为约12、13、14、15、16、17、18、19、20、21、22、23、24、25,26、27、28、29或30个核苷酸。优选地,第一和第二引物被设计成使得它们分别与第一和第二引物结合序列的至少18个连续核苷酸互补。
[0262]
如本文中所详述的,第二引物可以包含缀合到5'端处的核苷酸的亲和标签。可以缀合到5'端的核苷酸的任何亲和标签都适用于本发明的优选实施方案,其中将包含感兴趣的序列的反向互补的第二链或其部分与包含感兴趣的序列的第一链或其部分分离。
[0263]
替代5'端缀合标签,亲和标签可以位于第二引物序列的内部。例如,第二引物可以包含一个或多个生物素修饰的胸苷残基。
[0264]
如本文所用,术语“亲和标签”是指可以用于将附接有亲和标签的分子与不含有亲和标签的其他分子分离的部分。在某些情况下,“亲和标签”可以与“捕获剂”结合,其中亲和标签与捕获剂特异性结合,从而促进将附接有亲和标签的分子与不含有亲和标签的其他分子分离。亲和标签的实例包括6-组胺基嘌呤(6-histaminylpurine)(如例如在min和verdine,1996nucleic acids research 24:3806-381中所描述的)、多核苷酸尾,诸如能够附接到固体支持物的具有poly t互补的poly a尾、或能够例如与固体支持物上的抗生蛋白链菌素或抗生物素蛋白附接的生物素,其中生物素是最优选的。
[0265]
如本文所用,术语“生物素”是指包括生物素或生物素类似物(诸如双生物素、脱硫生物素、pc-生物素、氧代生物素、2'-亚氨基生物素、二氨基生物素、生物素亚砜、生物素叠氮化物、生物胞素等)的亲和剂,。优选地,生物素部分以至少10-8
m的亲和力与抗生蛋白链菌素结合。生物素亲和剂还可以包括接头,例如,-lc-生物素、-lc-lc-生物素、-slc-生物素或-pegn-生物素,其中n为3-12。
[0266]
在本发明的优选方法中,第二引物包含亲和标签。
[0267]
亲和标签可以至少存在于第二引物上。在本文中应进一步理解,亲和标签可以存在于第一引物和第二引物二者上。可选地,亲和标签不存在于第一引物上,例如,亲和标签仅存在于第二引物上。
[0268]
因此,核酸前体的扩增产生了包含至少一个标签的扩增的双链核酸前体,其中标签在包含与第一链互补的序列的链上。扩增的双链核酸前体还可以进一步在第一链上、优选在第一链的5'端处包含标签。第一链上的标签和第二链上的标签可以是相同或不同类型的标签。作为非限制性实例,第一链和第二链上的标签可以是生物素。
[0269]
在优选的实施方案中,核酸前体的扩增产生了包含标签的扩增的双链核酸前体,所述标签仅在包含与第一链互补的序列的链上。具体而言,包含与第一链互补的序列的链在5'端处包含标签。最优选地,互补链在5'端处包含生物素。
[0270]
可选地,例如当第二引物包含一个或多个生物素修饰的胸苷残基时,生物素部分可以例如存在于互补链的序列的内部。
[0271]
优选地,在结合固体支持物之前,将扩增的双链前体纯化。优选地,纯化导致了将扩增的和标签化的前体与(未使用)标签化的第二引物分离。双链前体的纯化可以使用本领
域已知的任何方法来进行,以纯化扩增的核酸产物。优选的纯化方法包括但不限于柱纯化(例如,qiaquick pcr纯化柱)以及在琼脂糖或丙烯酰胺凝胶上分离。
[0272]
消化
[0273]
本发明的方法包括一种用识别第一核酸内切酶识别位点的第一限制性或切口核酸内切酶以及用识别第二核酸内切酶识别位点的切口核酸内切酶来消化扩增的双链前体的步骤。用第一和第二核酸内切酶消化导致了通过在感兴趣的序列的紧接上游和下游切割糖-磷酸骨架来生产扩增的双链核酸前体。
[0274]
与第一核酸内切酶识别位点结合的第一核酸内切酶切割两个糖-磷酸骨架(作为限制性核酸内切酶),或者仅切割两个糖-磷酸骨架中的一个(作为切口核酸内切酶)。在第一核酸内切酶是切口核酸内切酶的情况下,第一核酸内切酶识别位点被定向成使得切口核酸内切酶在感兴趣的序列的紧接上游切割第一链。
[0275]
如本文中所指示的,与第一核酸内切酶识别位点结合的第一核酸内切酶优选地是外部切割器,例如在与核酸内切酶识别序列紧接(直接)相邻或在核酸内切酶识别序列之外的至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15或16个核苷酸处切割糖-磷酸骨架,如上文中所详述的。此类酶的实例是“iis型限制性酶”。第一核酸内切酶至少在感兴趣的序列的直接紧接上游(5
’
)切割糖-磷酸骨架。因此,第一核酸内切酶切割:i)第一dna链;或ii)第一和第二dna链。
[0276]
因此,第一核酸内切酶可以是切割dna两条链的外部切割器(即,限制性核酸内切酶)、或仅切割dna一条链的外部切割器(即,切口核酸内切酶)。在两种情况下,第一核酸内切酶识别位点被设计成使得外部切割器以如下定向结合位点:允许核酸内切酶在核酸内切酶识别位点的3
’
切割第一链的糖-磷酸骨架。更优选地,第一核酸内切酶识别位点被设计成使得外部切割器以如下定向结合位点:允许核酸内切酶在核酸内切酶识别位点的3
’
以及感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架。
[0277]
下文给出适合用作第一核酸内切酶的核酸内切酶的非限制性实例。
[0278]
适合用作第一核酸内切酶的切割两条dna链的核酸内切酶的非限制性实例是:mnli、bspcni、bsri、btsimuti、hphi、hpyav、mboii、acui、bcivi、bmri、bpmi、bpuei、bseri、bsgi、bsmi、bsrdi、btsαi、btsi、ecii、mmei、nmeaiii、asuhpi、bse1i、bsegi、bsemii、bseni、bsrsi、bstf5i、hin4ii、tscai、tsefi、tspdti、tspgwi、apypi、bce83i、bfii、bfui、bmui、bsami、bsbi、bscci、bse3di、bsemi、bsui、cchii、cchiii、cdpi、cjeniii、cstmi、eco57i、eco57mi、gsui、mva1269i、pcti、pladi、psppri、rdegbii、rleai、sdeai、taqii、tsoi、tth111ii、wvii、aquii、aquiv、drari、maqi、pspomii、rcei、rpab5i、rpabi、rpai、sste37i和rdegbiii。
[0279]
用作第一核酸内切酶的优选切口核酸内切酶可以选自:nt.alwi、nt.bsmai、nt.bstnbi和nt.bspqi(new england biolabs)。特别优选的第一核酸内切酶是nt.alwi。
[0280]
本领域技术人员了解如何选择第一核酸内切酶以及如何设计第一核酸内切酶识别位点,以确保核酸内切酶至少在感兴趣的序列的5'核苷酸紧接上游切割糖磷酸骨架。
[0281]
用识别第二核酸内切酶识别位点的第二核酸内切酶(第二核酸内切酶)来额外地消化扩增的双链前体。第二核酸内切酶可以是切割dna两条链的外部切割器(即,限制性核酸内切酶)、或仅切割dna一条链的外部切割器(即,切口核酸内切酶)。在两种情况下,第二
核酸内切酶识别位点被设计成使得外部切割器以如下定向结合位点:允许核酸内切酶在核酸内切酶识别位点的5
’
、紧接感兴趣的序列3
’
的最后核苷酸之后切割第一链的糖磷酸骨架。因此,第二核酸内切酶识别位点被设计成使得外部切割器以如下定向结合位点:允许核酸内切酶在核酸内切酶识别位点的5
’
以及感兴趣的序列的紧接下游切割第一链的糖磷酸骨架。
[0282]
如本文中所指示的,与第二核酸内切酶识别位点结合的第二核酸内切酶优选地是外部切割器,例如在与核酸内切酶识别序列紧接(直接)相邻或在核酸内切酶识别序列上游的至少1、2、3、4、5、6、7、8、9、10、11、12、13、14、15或16个核苷酸处切割糖-磷酸骨架,如上文中所详述的。在第二核酸内切酶是限制性核酸内切酶的情况下,它可以选自如本文上文中所指示的、与适合用作第一核酸内切酶的切割dna两条链的合适核酸内切酶相同的清单。
[0283]
如本文中所指示的,在特定实施方案中,优选的是,识别并结合第二核酸内切酶识别位点的第二核酸内切酶是切口核酸内切酶,即,核酸内切酶仅在感兴趣的序列的(末端)3'核苷酸的紧接下游切割双链dna的第一链。
[0284]
适合用作第二核酸内切酶的切口核酸内切酶可以选自:nb.bsrdi、nb.btsi、aspcni、bscgi、bspnci、fini、tsui、ubaf11i、bspgi、drdii、pfl1108i、ubapi、ecohi、unbi或vpac11ai。特别优选的第二核酸内切酶是nb.bsrdi。
[0285]
根据制造商说明书,在合适的温度,在合适的缓冲剂中,通过使(扩增的)前体与一种或多种酶接触来进行扩增的核酸前体的限制和/或切口。可以同时添加第一和第二核酸内切酶。可选地,可以使前体与第一(或第二)核酸内切酶接触,任选地将前体纯化,然后将第二(或第一)核酸内切酶添加到适当的缓冲剂中。在使用第一和第二核酸内切酶限制后,可以将所限制的前体纯化。
[0286]
固定
[0287]
在本发明方法的优选实施方案中,扩增的双链核酸前体的第二链包含与捕获剂接触的亲和标签,其中所述捕获剂优选地包含在固体支持物上。合适的捕获剂取决于亲和标签。例如,如果核酸包含生物素标签,则捕获剂可以是例如抗生蛋白链菌素或抗生物素蛋白。其他可能的标签可以是his标签、dnp(2,4-二硝基苯基)或地高辛配基(dig),其中捕获剂可以分别是抗his抗体、抗dnp抗体或抗dig抗体。类似地,如果亲和标签包含多核苷酸尾,则捕获剂可以是其互补序列。
[0288]
固体支持物或凝胶可以包含捕获剂。优选地,捕获剂存在于固体支持物上。因此,亲和标签与捕获剂的结合可以导致将扩增的标签化的双链核酸前体固定到固体支持物,和/或将标签化的单链寡核苷酸固定到固体支持物。适合于固定标签化的核酸的任何固体支持物都适用于本发明的方法。
[0289]
具有内表面或外表面的固体支持物可以是任何合适的形式,包括颗粒、粉末、片状物、珠、过滤器、平坦的基材、管状物、隧道状物、通道状物、金属颗粒等。支持物可以是多孔的,这可以提供用于使核酸前体的固定发生的内表面。优选的材料不会干扰标签化的核酸前体与捕获剂之间的相互作用。合适的材料可以包括但不限于纸、玻璃、陶瓷、金属、类金属、聚丙烯酰吗啉(polacryloylmorpholine)、各种塑料和塑料共聚物(诸如nylon
tm
、teflon
tm
、聚乙烯、聚丙烯、聚(4-甲基丁烯)、聚苯乙烯、聚苯乙烯、聚苯乙烯/乳胶、聚甲基丙烯酸酯、聚(对苯二甲酸乙二酯)、人造丝、尼龙、聚(丁酸乙烯酯)、聚偏二氟乙烯(pvdf)、硅
酮类、聚甲醛、纤维素、醋酸纤维素、硝化纤维素和可控孔径玻璃(controlled pore glass,inc.,fairfield,n.j.)、气凝胶等、以及通常已知适合用于亲和柱(例如,hplc柱)的任何材料。
[0290]
固体支持物可以是可单独或成组识别的珠(或具有合适表面的其他小物体)的形式。优选地,固体支持物也可以是根据其磁性性能而可分离的。因此,在本发明的优选实施方案中,亲和标签是生物素或包含生物素,并且固体支持物包含抗生蛋白链菌素。优选地,固体支持物是珠,并且其中更优选地,珠是磁性珠。特别优选的固体支持物是或类似材料。
[0291]
在特别优选的实施方案中,可以通过与功能化的(顺)磁性颗粒(或珠)温育来进行固定,其中颗粒是功能化的,原因在于它们的表面包含如本文中所定义的第二引物的标签的结合配偶体。在此种标签是生物素的情况下,可以用抗生蛋白链菌素将颗粒功能化。颗粒(或珠)的直径优选地为约1-5μm,并且可以包含以下特征中的一个或多个:亲水珠表面、基于羧酸的珠、直径为约1.05μm、等电点ph为5.2、中等荷电状态(-35mv(在ph7处)、铁含量(铁素体)为约26%(37%)、以及低聚集度。
[0292]
变性
[0293]
在本发明的优选实施方案中,将扩增的、并且优选地消化的双链核酸前体变性,例如,将第一链与第二互补链分离。本领域技术人员熟悉使双链dna变性的各种方法。此类方法可以包括但不限于将双链dna暴露于高温和/或化学物质。优选地,本发明方法中的变性包含化学变性。使dna变性的优选化学物质是例如甲酰胺、胍、水杨酸钠、二甲基亚砜(dmso)、丙二醇、尿素或碱性物质。优选地,化学变性是通过添加强碱来增加ph而进行的。优选地,强碱是碱金属类氢氧化物。具体而言,用于增加ph的合适强碱(或其组合)可以优选地选自naoh、lioh、koh、rboh、csoh、mg(oh)2、ca(oh)2、sr(oh)2和ba(oh)2。最优选地,在本发明的方法中,用于使双链核酸前体变性的强碱是碱金属类氢氧化物naoh。
[0294]
强碱可以优选地以约0.5-1.5m、优选约0.7-1.2m的终浓度添加,或者优选地,终浓度为约0.7、0.8、0.9、1.0、1.1或1.2m。最优选地,终浓度为约1m。
[0295]
可以将双链前体与强碱温育约1至30分钟、优选5至15分钟,或者优选地,将双链前体温育至少约1、2、3、4、5、6、7、8、9、10、11、12、13、14或15。最优选地,可以将双链前体与强碱温育约10分钟。
[0296]
在使双链前体变性后,可以添加酸以中和反应。该中和反应可以在如下文中所描述的将固体支持物与单链寡核苷酸分离之前或之后进行。优选地,中和反应在分离之后进行。任何酸都可以适用于中和。优选地,酸是强酸,诸如hcl、hi、hbr、hclo4、hno3或h2so4,其中hcl是最优选的。
[0297]
强酸优选地以约0.5-1.5m、或约0.7-1.2m的终浓度添加,或者优选地,终浓度为约0.7、0.8、0.9、1.0、1.1或1.2m。最优选地,终浓度为约1m。优选地,添加与用于变性的碱等摩尔量的酸,从而导致完全中和。
[0298]
分离
[0299]
在其中将包含感兴趣的序列的反向互补的第二链或其部分与包含感兴趣的序列的第一链或其部分分离的本发明优选方法包含一种将固体支持物去除以获得具有感兴趣的序列的单链寡核苷酸的步骤。
[0300]
固体支持物包含捕获剂。在本发明的方法中,捕获剂(例如,抗生蛋白链菌素)已经捕获了亲和标签(例如,生物素),并且亲和标签优选地偶联到核酸前体的互补(第二)链。因此,将固体支持物与单链寡核苷酸分离还必需将(标签化的)互补链与单链寡核苷酸分离。
[0301]
可以使用本领域已知的任何常规方法将固体支持物与单链寡核苷酸分离,并且该方法将取决于所使用的固体支持物的类型。例如,在固体支持物包含小颗粒的情况下,可以将这些颗粒离心,并且优选地将包含寡核苷酸的上清液转移到另一小瓶中。
[0302]
在固体支持物包含磁性或顺磁性珠的情况下,可以通过磁性分离(例如,通过将磁体放置在固体支持物附近)将固体支持物去除。
[0303]
纯化
[0304]
将固体支持物去除之后获得的单链寡核苷酸可以任选地进一步纯化。因此,在本发明的优选实施方案中,方法进一步包含纯化单链寡核苷酸的步骤g)。
[0305]
可以使用本领域已知的任何常规寡核苷酸纯化方法来进行纯化。优选的纯化方法是亲和纯化,诸如(微型)柱纯化。然而,其他纯化方法(例如,在琼脂糖或丙烯酰胺凝胶上分离)可以同样适合于纯化单链寡核苷酸。
[0306]
标记
[0307]
随后,可以对在本发明的方法中获得的单链寡核苷酸进行标记。例如,可以用荧光团、半抗原、亲和配体或放射性部分对生产的单链寡核苷酸进行标记。可选地,不对生产的单链寡核苷酸进行标记。
[0308]
如本文中所详述的,本发明特别适合于生产单链dna寡核苷酸。尽管如此,方法也可以产生rna分子(例如用于基因组编辑方法),诸如crispr-cas引导rna(如例如在mali等人,2013,nature methods,10(10):957-63和cong等人2013,science,339(9121):819-23中所描述的)。例如,为了生产rna分子,可以对本发明的方法进行如下修改:如本文中所详述的方法的步骤a)包含至少一种(单链或双链)核酸前体,其在5'至3'方向上包含以下元件:(1)第一引物结合位点,(2)感兴趣的序列,以及(3)第二引物结合位点。感兴趣的序列可以包含编码rna的序列,并且可以进一步包含用于转录rna的启动子,优选t7启动子。优选地,启动子可操作地链接感兴趣的序列。在得到步骤b)中的(任选地未标签化的)双链寡核苷酸之后,其中任选地,第二引物不包含标签。可以使用本领域已知的常规方法、诸如使用t7启动子(并且具有作为辅因子的mg
2+
)来从双链体dna转录rna。
[0309]
本发明的其他方面
[0310]
在第二方面,本发明涉及一种包含第一链的核酸前体,其中所述第一链在5'至3'方向上包含以下元件:
[0311]
(1)第一引物结合位点;
[0312]
(2)第一核酸内切酶识别位点;
[0313]
(3)感兴趣的序列;
[0314]
(4)第二核酸内切酶识别位点;以及
[0315]
(5)第二引物结合位点。
[0316]
优选地,如本发明的第一方面中进一步详述的,第一引物可以选择性地仅与第一引物结合序列退火,并且如本发明的第一方面中进一步详述的,第二引物可以选择性地仅与第二引物结合序列退火。任选地,第一和第二引物以及第一和第二引物结合位点可以相
同或相似,这样第一引物与第二引物结合位点退火并且反之亦然,以允许对核酸前体进行扩增。
[0317]
优选地,第一核酸内切酶识别位点被设计成使得在双链化之后,第一核酸内切酶在感兴趣的序列的紧接上游切割第一链的糖-磷酸骨架。
[0318]
优选地,第二核酸内切酶识别位点被设计成使得在双链化之后,切口核酸内切酶在感兴趣的序列的紧接下游切割第一链的糖-磷酸骨架。
[0319]
优选地,前体被设计成使得感兴趣的序列的糖-磷酸骨架(即,从感兴趣的序列的5'核苷酸至感兴趣的序列的3'核苷酸)不被本发明方法中使用的第一和第二核酸内切酶切割。
[0320]
优选地,感兴趣的序列不包含第一和第二核酸内切酶识别位点或其反向互补。
[0321]
核酸前体可以是单链或双链核酸前体。如果核酸前体是双链的,则前体包含与第一链互补的第二链。如本文上文中所详述的,进一步指定了前体。在最优选的实施方案中,核酸前体具有选自seq id no:1-978的序列。
[0322]
核酸前体可以是双链的。在进一步优选的实施方案中,双链核酸前体包含亲和标签。
[0323]
优选地,亲和标签位于第二链的5'端处。例如,互补链的5'核苷酸可以包含生物素标签或多核苷酸尾。优选地,互补链在第二链的5'端处包含生物素标签,即在5'端处被生物素化。可以使用本领域已知的任何常规方法将生物素部分缀合到5'核苷酸。
[0324]
可选地,亲和标签位于互补序列内部。优选地,此种内部亲和标签位于第二链第二核酸内切酶识别位点的5
’
(即,与第一链的核酸内切酶识别位点反向互补的序列的5')上。更优选地,此种内部亲和标签位于第二链第二引物结合位点处(即,与第一链的第二引物结合序列反向互补的序列上)。此种内部亲和标签的优选实例是生物素修饰的胸苷残基。
[0325]
优选地,双链核酸前体在第一链的3'端和/或5'端处不包含亲和标签。优选地,双链核酸前体仅在第二链的5'端处包含亲和标签。
[0326]
在第三方面,本发明涉及一种固体支持物,其包含如本文上文中所定义的双链核酸前体。如本文上文中所详述的,进一步指定了固体支持物。优选地,通过亲和捕获将双链核酸前体结合到固体支持物。双链核酸前体的第一链和第二链可以具有完全完整的糖-磷酸骨架。可选地,前体的第一链可以包含至少一个或两个磷酸二酯键切割,并且前体的第二链具有完全完整的糖-磷酸酯骨架,或者可选地,前体的第一链可以包含至少一个或两个磷酸二酯键切割,并且前体的第二链具有至多一个磷酸二酯键切割。
[0327]
在进一步的实施方案中,固体支持物包含单链第二链,即,与如本文上文中所定义的第一链互补的链。
[0328]
在第四方面,本发明涉及一种试剂盒,其含有用于本发明方法的元件。此种试剂盒可以包含在其中容纳一个或多个容器的载体,诸如管或小瓶。
[0329]
优选地,试剂盒包含以下各项中的至少一个:
[0330]-容器(1),其包含第二(切口)核酸内切酶和任选地包含如本文上文中所定义的第一核酸内切酶;
[0331]-容器(2),其包含用于如本文上文中所定义的扩增步骤的酶;
[0332]-容器(3),其包含用于如本文上文中所定义的亲和纯化的固体支持物;以及
[0333]-容器(4),其包含用于如本文上文中所定义的变性的化学制品。
[0334]
在优选的实施方案中,试剂盒包含容器(1)和(2)、或(1)和(3)、或(1)和(4)。在另一优选的实施方案中,试剂盒包含容器(2)和(3)、或(2)和(4)、或(3)和(4)。在另一优选的实施方案中,试剂盒包含容器(1)、(2)和(3);或(1)、(2)和(4);或(1)、(3)和(4)。在另一优选的实施方案中,试剂盒包含容器(2)、(3)和(4);或(1)、(2)、(3)和(4)。在最优选的实施方案中,试剂盒包含容器(1)、(2)、(3)和任选地包含容器(4)。
[0335]
在进一步优选的实施方案中,如上文中所定义的试剂盒进一步包含容器(5),所述容器(5)包含如本文上文中所定义的第一和/或标签化的第二引物。可选地,第一和/或第二标签化的引物可以包含在包含用于扩增步骤的酶的容器(2)内。
[0336]
试剂可以以冻干形式存在,或者存在于适当的缓冲剂中。试剂盒还可以含有用于执行本发明所需的任何其他组分,诸如缓冲剂、移液管、微量滴定板和书面说明。用于本发明试剂盒的此类其他组分是本领域技术人员已知的。
[0337]
在第五方面,本发明涉及如本文中所定义的核酸前体或如本文中所定义的试剂盒套装用于生产一个或多个单链寡核苷酸的用途。生产的单链寡核苷酸可以包含如本文上文中所定义的感兴趣的序列或由如本文上文中所定义的感兴趣的序列组成。
[0338]
在第六方面,本发明涉及如本文中所定义的核酸前体或如本文中所定义的试剂盒套装用于扩增一个或多个单链寡核苷酸的用途。生产的单链寡核苷酸可以包含如本文上文中所定义的感兴趣的序列或由如本文上文中所定义的感兴趣的序列组成。
附图说明
[0339]
图1:本发明方法的优选实施方案的示意图。pbs1是第一引物结合位点,pbs2是第二引物结合位点,es1是第一核酸内切酶识别位点,并且es2是第二核酸内切酶识别位点。反向引物可以包含标签(黑色圆圈)。固体支持物(大圆圈)可以捕获标签化的、扩增的和切口的核酸前体。
[0340]
图2:本发明的两个例示性核酸前体。a)第一核酸内切酶识别位点可以(部分或完全地)包含在第一引物结合位点内,并且第二核酸内切酶识别位点可以(部分或完全地)包含在第二引物结合位点内。b)核酸前体,其中元件是五个不同的元件。缩写和符号如图1所指示。箭头表示引物,并且反向引物可以包含标签(黑色圆圈)。
[0341]
图3:本发明的例示性核酸前体。引物结合位点(pbs)可以与核酸内切酶识别位点(es,黑色)重叠。另外,引物结合位点可以包含通用部分(黑色和白色)和可变部分(灰色)。a)使用与引物结合位点的通用部分和可变部分互补的引物对的扩增允许对核酸前体的特异性亚组进行扩增。b)使用仅与通用部分互补的引物对的扩增允许对核酸前体的整个库进行扩增。缩写和符号如图1和图2所指示。
[0342]
图4:本发明的例示性核酸前体。核酸前体可以包含五个不同的元件。引物结合位点可以包含通用部分(白色)和可变部分(灰色)。a)使用与引物结合位点的通用部分和可变部分互补的引物对的扩增允许对核酸前体的特异性亚组进行扩增。b)使用仅与通用部分(白色)互补的引物对的扩增允许对核酸前体的整个库进行扩增。缩写和符号如图1和图2所指示。
[0343]
图5:本发明的例示性核酸前体。引物结合位点(pbs)可以与核酸内切酶识别位点
(es,黑色)重叠。引物结合位点可以包含可变部分(灰色)和通用部分(黑色和白色)。a)使用仅与可变部分和es完全互补的引物对的扩增允许对核酸前体的特异性亚组进行扩增。b)使用仅与通用部分(白色)互补的引物对的扩增允许对核酸前体的整个库进行扩增。缩写和符号如图1和图2所指示。
[0344]
图6:本发明的例示性核酸前体。核酸前体可以包含五个不同的元件。引物结合位点可以包含可变部分(灰色)和通用部分(白色)。a)使用仅与可变部分完全互补的引物对的扩增允许对核酸前体的特异性亚组进行扩增。b)使用仅与通用部分(白色)互补的引物对的扩增允许对核酸前体的整个库进行扩增。缩写和符号如图1和图2所指示。
[0345]
图7:结果tapestation d1000(agilent):检查了200μl未纯化pcr总样本中的1μl样本,并且检查了50μl(纯化)rpa总样本中的1μl样本。
[0346]
图8:结果tapestation d1000:检测到清晰可见的102bp双链扩增产物(检查了100μl总数中的1μl),产物预期是扩增的探针前体。尺寸差异很可能是由于不正确地定型(sizing)tapestation系统所致。
[0347]
图9:用生物素纯化,结果agilent small rna试剂盒(检查了40μl总数中的1μl的1/4稀释样本)。回收的dna与预期的单链55-63nt探针相对应。尺寸差异很可能是由于不正确地定型阵列系统所致。
[0348]
图10:结果small rna agilent比较实验
[0349]
实施例
[0350]
使用包含pcr扩增、扩增子切口、通过丙烯酰胺-凝胶分离来纯化切口的扩增子、以及随后进行热变性以释放探针的方法对多重9个探针前体进行探针扩增的初步实验未导致令人满意的探针产量。使用生物素-珠纯化代替丙烯酰胺-凝胶分离,与化学变性而不是热变性组合,克服了该问题。然而,将多重水平提高到3912个探针又导致了低产量和异源双链体形成(参见实施例1)。通过使用等温扩增方法代替pcr、连同使用用于扩增子纯化的生物素-珠以及用于探针释放的化学变性,克服了这些问题。导致高产量且没有异源双链体形成的扩增方法在实施例2和3中进行了详细描述。
[0351]
实施例1.用于高度多重探针的pcr和rpa的比较
[0352]
探针前体
[0353]
将3912个探针前体(平均长度为90nt)(包含978个独特序列;seq id no:1-978)在lc sciences的可编程微阵列上合成。将25μl的无核酸酶水添加到冻干样本中,使其浓度为0.064pmol/μl。
[0354]
探针前体的处理
[0355]
pcr:
[0356]
在200μl总体积中进行pcr扩增,总体积含有0.05pmol多重探针前体(总量)、200μm dntp、4μm f-引物(seq id no:979)、4μm r-生物素-引物(seq id no:no:980)(未生物素化的引物的序列在seq id no:981中给出)、1x cloned pfu反应缓冲剂_ad(agilent)中的10个单位克隆pfu dna聚合酶_ad。使用以下pcr程序:在95℃5分钟,然后在95℃进行30秒的二十个循环,在55℃2分钟,在72℃八分钟,然后在72℃10分钟。rpa:
[0357]
使用twistdx的twistamp basic试剂盒(order#tabas01kit)进行重组酶聚合酶扩增(rpa)。制备反应混合物,反应混合物含有0.05pmol多重探针前体(总量)、700nm f-引物
(seq id no:979)、700nm r-生物素-引物(seq id no:980)和29.5μl再水化缓冲剂。向反应混合物添加mq至终体积为47.5μl。在添加2μl的280mm mgac以开始反应之后,将混合物在38℃温育40分钟。
[0358]
根据制造商的方案,用qiaquick pcr纯化柱纯化样本,并且使用50μl eb缓冲剂洗脱。
[0359]
结果:
[0360]
用agilent d1000 screen tape在tapestation上检查分别通过pcr和rpa产生的扩增子的质量和尺寸(图7)。pcr产生的特异性扩增子产量低(与rpa相比),这可能是由于异源双链体形成所致。
[0361]
实施例2.用于探针扩增和纯化的方法
[0362]
探针前体
[0363]
将3912个探针前体(平均长度为90nt)(包含978个独特序列;seq id no:1-978)在lc sciences的可编程微阵列上合成。将25μl的无核酸酶水添加到冻干样本中,使其浓度为0.064pmol/μl。
[0364]
探针前体的处理
[0365]
使用twistdx的twistamp basic试剂盒(order#tabas01kit)进行重组酶聚合酶扩增(rpa)。制备单一rpa反应混合物,反应混合物含有0.01pmol多重探针前体(总量)、700nm f-引物(seq id no:979)、700nm r-生物素-引物(seq id no:980)和29.5μl再水化缓冲剂。向反应混合物添加mq至终体积为47.5μl。将反应混合物添加到冷冻干燥的碱性反应中。在添加2μl的280mm mgac以开始反应之后,将混合物在38℃温育40分钟。
[0366]
进行八个单独的rpa反应并合并。根据制造商的方案,使用两个qiaquick pcr纯化柱纯化扩增子,并且每柱使用50μl eb缓冲剂洗脱,即,总计100μl eb缓冲剂。
[0367]
用agilent d1000 screen tape在tapestation上检查扩增子的质量和尺寸(图8)。用life technologies的qubit dsdna br测定试剂盒(cat#q32850)测量浓度(表1)。总产量约为8μg扩增子。
[0368]
表1:结果qubit(检查了总计100μl中的1μl)
[0369][0370]
单链55-63nt.靶向探针的切口
[0371]
(85-93nt.)探针前体的侧接序列含有识别位点,所述识别位点用于在与靶向臂的连接处的切口限制性核酸内切酶。
[0372]
进行如下两个切口反应:将50μl柱纯化的rpa反应液、10μl 10x cut-smart缓冲剂(new england biolabs)、5μl nt.alwi(10u/μl,new england biolabs)和35μl mq混合并在37℃温育两小时。在该步骤之后,添加5μl的nbbsrdi(10u/μl,new england biolabs)并在65℃温育两小时,然后在80℃进行20分钟的失活步骤。
[0373]
将两个反应的切口rpa产物合并,并且根据制造商的方案,使用两个qiaquick pcr纯化柱进行纯化,每柱用80μl eb缓冲剂进行洗脱(总计160μl)。
[0374]
用生物素纯化
[0375]
根据制造商的方案,使用dynabeads myone抗生蛋白链菌素c1(cat#65002)来固定qiaquick纯化的切口rpa产物。将160μl qiaquick纯化产物分成三个53.3μl的等分试样。将量为200μl的珠添加到这些等分试样的每一个中。根据制造商的方案,进行温育,并且进行洗涤。在最后步骤,将珠再悬浮于每等分试样20μl eb缓冲剂中。
[0376]
单链55-63nt.靶向探针的释放
[0377]
将上文中获得的三个等分试样中的每一个均进行化学变性。为了进行化学变性,添加naoh至终浓度为0.9m。将混合物在室温温育10分钟,然后放置在磁体上。将上清液取出,并且通过添加与所添加的naoh等摩尔量的hcl来中和。
[0378]
将三个等分试样的上清液合并,并且根据制造商的方案,使用zymo research(cat#d7010)的ssdna/rna clean&concentrator进行纯化。用40μl eb(qiagen)进行洗脱。
[0379]
使用相当长度(54-68nt)的有序探针组作为阳性对照,用agilent small rna试剂盒在生物分析仪上检查探针的质量和尺寸(图9)。用life technologies的qubit ssdna测定试剂盒(cat#q10212)测量浓度(表2)。
[0380]
表2:结果qubit
[0381][0382]
结果
[0383]
本探针扩增方法导致了以非常低量的输入材料(0.01pmol)实现高的净探针产量(探针产量的净倍数增加为550)。该方法允许以高度多重水平扩增寡核苷酸而不产生异源双链体分子。使用生物素珠进行纯化提供了一种非常快速简便的方法。进一步地,用于释放扩增的寡核苷酸的化学变性和中和作用非常有效,而使用加热进行变性和释放不会生产可检出量的产物。
[0384]
实施例3.参数变化
[0385]
在一组比较实验中,改变一个时间参数,进行实施例2中详细描述的方法。实验设计如下:
[0386]
1.如实施例2中所详述的方法,但是每种切口酶使用2.5μl(每种12.5个单位),而不是如实施例2中所使用的5μl(每种50个单位)(图10“两种切口酶”)。
[0387]
2.如实施例2中所详述的方法,其中以与如在1下所指示的相同的体积和单位,用alwi(new england biolabs)代替切口酶nt.alwi(图10:“一种限制酶和一种切口酶”)。
[0388]
用agilent small rna试剂盒在生物分析仪上检查探针的质量和尺寸(图10)。用限制酶代替第一切口酶获得相当的产量。
[0389]
本领域技术人员理解,尽管本文中指定的实验涉及用作探针的寡核苷酸,但是相同的方案适用于意图用于不同用途的寡核苷酸。
[0390]
实施例4.扩增的寡核苷酸探针验证
[0391]
在使用如实施例2中所详述的方法中生产的3912个寡核苷酸探针被设计成在ola测定中检测玉米(玉蜀黍(zea mays))基因组中的326个不同snp,每个具有2个等位基因(即326重)。如实施例2中所生产的探针通过在ola测定中测试对5个不同的基因组玉米dna样本进行基因分型来验证,这些样本是由f2玉蜀黍定位群体制备的。更具体地,通过对5个不同基因组玉米dna样本中的每一个的重复(duplicate)之间的基因型识别(calling)进行比较,测试使用这些探针的ola测定的再现性。进一步地,通过将这些5个不同样本内的基因型识别与使用相同ola测定和相同5个不同基因组玉米dna样本的基因型识别进行比较,验证使用这些探针的ola测定,其中这些探针被现有1056重ola测定(idt,integrated dna technologies)单独合成的探针替代,现有1056重ola测定包含用于检测326个基因座的snp等位基因的326重探针。
[0392]
基于基因座的已知序列,使用普通程序设计寡核苷酸探针(5'-3'方向),并且加以选择以对326个基因座中的每一个的snp等位基因进行区分。包括pcr引物结合区域、基因座和等位基因标识符。更具体地,第一引物结合序列(长度为16个核苷酸)的反向互补位于等位基因特异性探针的5'端处,并且第二引物结合序列(长度为18个核苷酸)位于基因座特异性探针的3'端处。与第一引物结合序列的3'端相邻的是(在5'至3'方向上)以下元件:13个核苷酸的通用序列、4-碱基等位基因标识符、以及第一靶特异性序列。与第二引物序列的5'端相邻的是(在3'至5'方向上)以下元件:14个核苷酸的通用序列、8-碱基基因座标识符、以及第二靶特异性序列。
[0393]
下文中,使用如实施例2中所制备的探针对ola测定的过程进行描述。对于单独合成的探针,整个过程进行的完全相同,其中在连接反应中,如实施例2中所生产的1μl 326重探针混合物(每基因座3.4nm;总计1.12μm)被从idt订购的1μl 1056重探针混合物替代,随后进行磷酸化(每基因座0.4nm;总计0.4μm)。
[0394]
ola测定过程
[0395]
连接反应制备如下:将5μl中的100至200ng基因组dna与1μl10xtaq dna连接酶缓冲剂(200mm tris-hci ph 7.6、250mm kac、100mm mgac、10mm nad、100mm二硫苏糖醇、1%triton-x100)、4个单位taq dna连接酶(new england biolabs)、如实施例2中所生产的1μl 326重探针混合物(每基因座3.4nm;总计1.12μm)或从lc sciences订购的1μl 1056重探针混合物进行组合,随后进行磷酸化(每基因座0.4nm;总计0.4μm),并且添加milliq水至总量为10μl。以每个基因组dna样本一式四份设置连接反应。将反应混合物在94℃温育1分和30秒,随后每30秒温度降低1.0℃直到60℃,然后在60℃温育大约18小时。将反应保持在4℃直到进一步使用。将连接反应物用milliq水稀释4倍。
[0396]
使用第一和第二扩增引物对连接产物进行扩增。第一扩增引物被设计成包含在其3'末端处的用于与第一引物结合序列退火的序列(16个核苷酸)、位于其5'末端处的p7序列、以及在这些元件之间的5-碱基样本标识符。第二扩增引物被设计成包含在其3'末端处的用于与第二引物结合序列退火的序列(18个核苷酸)、位于其5'末端处的p5序列、以及在这些元件之间的6-碱基平板标识符。
[0397]
连接产物的扩增在以下反应混合物中进行:10μl 4x稀释的连接反应液,每种引物(第一和第二扩增引物)0.05μm(终浓度),20μl phusion hot start flx master mix(biok
é
),并且添加milliq水至总量为40μl。将每一连接产物扩增三次;每5个不同的基因组dna样
本,总计进行60次pcr反应。使用以下条件,在具有金或银单元(block)的pe9700(perkin elmer corp.)上进行热循环过程:步骤1:pcr预温育:在98℃30秒。步骤2:变性:在98℃10秒;退火:在65℃15秒。延伸:在72℃15秒。总循环数为29。步骤3:在72℃延伸5分钟。将反应保持在4℃直到进一步使用。将总计60次pcr反应的扩增产物合并(60x40μl),并且使用两个pcr纯化柱(qiagen)纯化,并且每柱用15μl miiliq水洗脱,总计30μl。
[0398]
用sage science的pippin prep对扩增子进行纯化。使用3%的盒以及标志物c纯化四次900ng,无溢出。洗脱范围为170bp直到230bp。使用minelute试剂盒(qiagen)纯化洗脱产物,并且用15μl洗脱。
[0399]
使用illumina miseq nano run对扩增子进行测序。对所得测序数据进行去多重化(de-multiplexed),其中将读段分配给所使用样本中的每一个。为了有效基因分型所需的足够的基因组覆盖度,将每个基因组dna样本的两个一次四份的数据合并,并且进行进一步的处理,并且被认为是单峰(singlet),从而导致每个基因组dna样本的重复结果。
[0400]
结果
[0401]
对于总计5个样本(包含理论总数为5x326=1630个基因型),总计识别1452个基因型,其中重复之间的再现性为99.8%,即,用如实施例2中所生产的探针使用326重测定识别的基因型中的99.8%在重复之间是相同的。当使用单独合成的探针时,总计识别1452个基因型,这些基因型与使用实施例2中生产的探针识别的基因型97.5%相同。
[0402]
表x:使用5个玉米基因组dna样本的326重ola测定的性能(基因型的理论总数为1630)
[0403][0404]
1)
识别的基因型与在使用单独合成的探针的ola测定中识别的基因型相匹配的百分比。
[0405]
2)
识别的基因型在重复之间相匹配的百分比。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签:

 热门咨询
热门咨询




tips