
 商标分类
商标分类  商标转让
商标转让 
一种β-石竹烯衍生的二氟烷基取代三环化合物的制备方法与流程
 2021-02-02 04:02:13|
2021-02-02 04:02:13| 345|
345| 起点商标网
起点商标网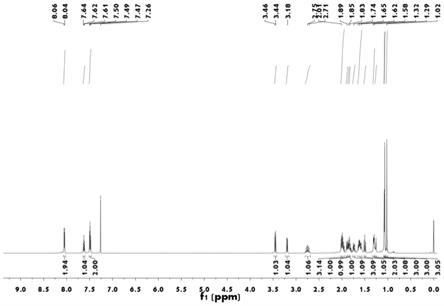
一种
β-石竹烯衍生的二氟烷基取代三环化合物的制备方法
技术领域
[0001]
本发明涉及有机合成领域,特别涉及一种β-石竹烯衍生的二氟烷基取代三环化合物的制备方法。
背景技术:
[0002]
β-石竹烯是一种具有四九反式并环结构双环倍半萜类化合物,是油樟油、丁香叶油、丁香茎油、肉桂叶油等的重要组成成分。β-石竹烯本身具有较好的生理活性,对结肠炎症细胞、骨肉瘤细胞具有很好的抑制效果(pan,z.,et al.bangladesh journal of pharmacology,2016,11,817.);也能够与阿霉素组合用药来增强药效减少对正常细胞的毒性(giacomo sd.,et al.anticancer reaserch.2017.37.1191-1196)。
[0003]
同时,在药物化学领域,对生理活性分子进行氟原子或含氟基团取代开发新的抗癌药物、抗肿瘤药物、抗病毒试剂、消炎药物、中枢神经系统药物的重要方法。其中,二氟亚甲基是一类特殊的基团,是醚氧的“等极体”和“等体体”。例如用二氟亚甲基取代磷酸酯中的氧原子,能够使母体分子的活性不会改变,但其稳定性得到了很大的加强。所以发展一种向β-石竹烯中引入含氟原子或含氟基团的方法具有重要意义。
[0004]
目前,向β-石竹烯中引入含氟原子或含氟基团的方法还没有任何报道。仅报道了几种β-石竹烯的官能团转化方法,具体合成实例如下:
[0005]
(一)在2010年,andrew l.lawrence等以β-石竹烯、2,4,6-三羟基-1,3-苯二醛及脂肪族或芳香族醛为原料通过一个hetero-diels-alder反应合成了具有生理活性的guajadial和psidial及其类似物(org.lett.2010,12,1676)。
[0006][0007]
(二)最近,南京林业大学徐莉等通过儿萘酚硼烷、氢氧化钾、双氧水对β-石竹烯九环中的环上双键进行选择性硼氢化氧化在九环上引入了羟基,随后通过酯化反应将呋喃甲酸基团与β-石竹烯进行了结合,经过no抑制率实验、细胞毒性实验和抗癌活性实验证实该类化合物对炎症具有一定的抑制效果,且对宫颈癌、肝癌、乳腺癌或肺癌均有很好的抗癌活性,可以应用于抗炎、抗癌症药物的制备中(cn 110804033 a)。
[0008][0009]
综上所述,β-石竹烯衍生物一般都具有较好的生理活性,氟原子或含氟基团取代
的β-石竹烯衍生物应该是一类潜在的生理活性分子。
技术实现要素:
[0010]
本发明正是针对以上技术问题,提供一种β-石竹烯衍生的二氟烷基取代三环化合物的制备方法。该方法用于生产β-石竹烯衍生的二氟烷基取代三环化合物生产成本低,工艺简便、生产安全可靠、绿色环保。
[0011]
为了实现以上发明目的,本发明的具体技术方案如下:
[0012]
一种β-石竹烯衍生的二氟烷基取代三环化合物的制备方法,其包括以下步骤:在惰性气体保护下,以常见极性试剂为反应溶剂,β-石竹烯和碘二氟甲基芳基酮或碘二氟乙酸酯为原料,碱、芳基酚和蓝光照射下,在一定温度下进行充分搅拌反应,反应达到终点后,分离、提纯得到二氟烷基取代三环化合物。
[0013]
碘二氟甲基芳基酮是指苯环或萘环或杂环上被甲基、甲氧基、氯、溴、碘、氟、硝基、氰基、乙基、异丙基一元或多元取代的碘二氟甲基芳基酮。例如:2,2,-二氟-2-碘-苯乙酮、2,2,-二氟-2-碘-对甲苯乙酮、2,2,-二氟-2-碘-对甲氧基苯乙酮、2,2,-二氟-2-碘-对氟苯乙酮、2,2,-二氟-2-碘-对溴苯乙酮、2,2,-二氟-2-碘-对氯苯乙酮、2,2,-二氟-2-碘-对硝基苯乙酮、2,2,-二氟-2-碘-对氰基苯乙酮、2,2,-二氟-2-碘-间甲苯乙酮、2,2,-二氟-2-碘-间甲氧基苯乙酮、2,2,-二氟-2-碘-间氟苯乙酮、2,2,-二氟-2-碘-间溴苯乙酮、2,2,-二氟-2-碘-间氯苯乙酮、2,2,-二氟-2-碘-间硝基苯乙酮、2,2,-二氟-2-碘-邻甲苯乙酮、2,2,-二氟-2-碘-邻甲氧基苯乙酮、2,2,-二氟-2-碘-邻氟苯乙酮、2,2,-二氟-2-碘-邻溴苯乙酮、2,2,-二氟-2-碘-邻氯苯乙酮、2,2,-二氟-2-碘-邻硝基苯乙酮、2,2,-二氟-2-碘-1-萘乙酮、2,2,-二氟-2-碘-2
’-
呋喃乙酮、2,2,-二氟-2-碘-2
’-
噻吩乙酮等。碘二氟乙酸酯是指含有甲基或乙基或叔丁基的碘二氟乙酸衍生物。如碘二氟乙酸甲酯、碘二氟乙酸乙酯、碘二氟乙酸叔丁酯等。
[0014]
作为本申请中的一个较好的实施方式,所述惰性气体为氮气或氩气;所述惰性气体保护采用储气袋通入反应体系。
[0015]
作为本申请中的一个较好的实施方式,所述常见溶剂为乙醇、叔丁基甲基醚、四氢呋喃、n,n-二甲基甲酰胺、n-甲基吡咯烷酮、二氯乙烷、1,4-二氧六环和乙腈中的至少一种。
[0016]
作为本申请中的一个较好的实施方式,所述碱为碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾、醋酸钠、醋酸钾、氢化钠、叔丁醇钾、叔丁醇钠、三乙胺和乙基二异丙基胺中的任意一种。
[0017]
作为本申请中的一个较好的实施方式,所述酚为苯酚、邻甲基苯酚、间甲基苯酚、对甲基苯酚、邻氯苯酚、间氯苯酚、对氯苯酚、邻溴苯酚、间溴苯酚、对溴苯酚、邻氟苯酚、间氟苯酚、对氟苯酚、a-萘酚和β-萘酚中的任意一种。
[0018]
作为本申请中的一个较好的实施方式,所述蓝光的波长范围为400nm-500nm。
[0019]
作为本申请中的一个较好的实施方式,β-石竹烯和碘二氟甲基芳基酮或碘二氟乙酸酯物质的量之比为1:1.0~5,β-石竹烯物质的量与反应溶剂体积之比为1:1~20moll-1
,该投料比和溶剂用量经济合算、节约成本。
[0020]
作为本申请中的一个较好的实施方式,反应终点由薄层色谱法检测反应液中原料β-石竹烯转化完全进行判定,薄层色谱硅胶为:硅胶gf254,薄层色谱展开剂为:石油醚∶乙
酸乙酯=1~1000∶1(体积比),显色方式:碘显色或者紫外显色(254nm),反应终点判断方法简便易行。
[0021]
作为本申请中的一个较好的实施方式,反应温度为0℃~120℃,反应温度可控,易于操作。
[0022]
作为本申请中的一个较好的实施方式,分离、提纯步骤为:反应达终点后,加入适量水猝灭反应,再加入乙酸乙酯萃取水相,合并有机相,有机相依次用nacl水溶液洗涤,无水硫酸钠干燥,过滤,旋转蒸发回收溶剂,残留物经重结晶或用硅胶柱层析分离得到β-石竹烯衍生的二氟烷基取代三环化合物。
[0023]
与现有技术相比,本发明的有益效果是:
[0024]
(一)、原料廉价易得,成本低。
[0025]
(二)、反应条件温和,可操作性强,安全性高,绿色环保,而且反应转化率和收率高。
[0026]
(三)、工艺流程短,反应规模易于扩大,产物分离简单,具有适于工业化生产的优势。
附图说明
[0027]
图1为实施例1得到的β-石竹烯衍生的二氟烷基取代三环化合物的1h nmr图。
[0028]
图2为实施例1得到的β-石竹烯衍生的二氟烷基取代三环化合物的
13
cnmr图。
[0029]
图3为实施例1得到的β-石竹烯衍生的二氟烷基取代三环化合物的高分辨谱图。
具体实施方式
[0030]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。此外应理解,在阅读了本发明的内容之后,本领域技术人员可以对本发明作各种改动或修改,但这些等价形式同样落于本申请所附权利要求书所限定的范围。
[0031]
极性溶剂便宜易得,作为反应溶剂,有助于原料的溶解,并能在有效稳定二氟烷基自由基,有效避免副反应及提高反应产率;1,4-二氧六环和n-甲基吡咯烷酮(nmp)为常用非质子极性溶剂,因此以下实施例中均使用无水1,4-二氧六环为反应溶剂。
[0032]
以下反应终点由薄层色谱法检测反应液中原料β-石竹烯转化完全进行判定,薄层色谱硅胶为:硅胶gf254,薄层色谱展开剂为:石油醚∶乙酸乙酯=1~100∶1(体积比),显色方式:碘显色或者紫外显色(254nm),反应终点判断方法简便易行。
[0033]
实施例1 β-石竹烯衍生的二氟苯乙酮及碘取代的三环化合物3a
[0034]
(2,2-difluoro-3-((2ar,7bs)-5-iodo-2,2,4a-trimethyldecahydro-7ah-cycl obuta[e]inden-7a-yl)-1l3-propan-1-one
--
argon(1/1))的制备:
[0035]
[0036]
向一个干燥无氧氮气保护的三口瓶中依次加入无水碳酸氢钾(0.6g,6mmol),2-苯酚(6mg,0.06mmol),1,4-二氧六环30ml,β-石竹烯(610mg,3mmol),碘二氟苯乙酮(1.69g,6mmol)。随后50℃反应瓶被置于蓝光照射下进行充分搅拌。通过薄层色谱法确认反应终点后,将反应瓶冷却至室温,加入适量水溶液进行猝灭反应,再加入乙酸乙酯萃取水相,合并有机相,有机相用饱和nacl水溶液洗涤,无水硫酸钠干燥,过滤,旋转蒸发回收溶剂,残留物用石油醚:乙酸乙酯=1~1000∶1(体积比)柱层析后,得到黄色固体1.38g,产率为95%。
[0037]1h nmr(600mhz,cdcl3)δ8.08(d,j=8.0hz,2h),7.62(t,j=7.4hz,1h),7.49(t,j=7.8hz,2h),3.40(d,j=9.9hz,1h),3.36(d,j=9.9hz,1h),2.75
–
2.63(m,1h),1.98
–
1.89(m,2h),δ1.88
–
1.82(m,1h),1.80(m,1h),1.76
–
1.71(m,2h),1.71
–
1.65(m,1h),1.62(dd,j=10.2,7.3hz,2h),1.59
–
1.54(m,1h),1.52
–
1.44(m,2h),1.25(s,3h),1.04(s,3h),1.01(s,3h).
13
c nmr(151mhz,cdcl3)δ189.68(t,j=30.9hz),134.15,132.35,130.20,128.71,120.97(t,j=256.4hz),53.50
–
52.08(t,j=20.4hz),49.60,46.58,40.72,40.18,39.72,37.91,36.32,30.57,28.33,24.39(d,j=3.2hz),23.98,20.44,17.94,17.04.ir(kbr)
max 3023,2914,1706,1654,1601,1475,1344,1271,1239,1175,1086,970,747,665cm-1
;ms(ei)m/z 486.1[m]
+
;hrms(ei)m/z[m]
+
calcd for c
23
h
29
of2i,486.1222;found,486.1228.
[0038]
实施例2 β-石竹烯衍生的二氟乙酸乙酯及碘取代的三环化合物3b
[0039]
(2,2-difluoro-1-((2ar,7bs)-5-iodo-2,2,4a-trimethyldecahydro-7ah-cycl obuta[e]inden-7a-yl)pentan-3-one)的制备:
[0040][0041]
向一个干燥无氧氮气保护的三口瓶中依次加入无水碳酸氢钾(0.6g,6mmol),2-苯酚(6mg,0.06mmol),1,4-二氧六环30ml,β-石竹烯(610mg,3mmol),碘二氟乙酸乙酯(1.50g,6mmol)。随后反应瓶被置于蓝光照射下50℃进行充分搅拌。通过薄层色谱法确认反应终点后,将反应瓶冷却至室温,加入适量水溶液进行猝灭反应,再加入乙酸乙酯萃取水相,合并有机相,有机相用饱和nacl水溶液洗涤,无水硫酸钠干燥,过滤,旋转蒸发回收溶剂,残留物用石油醚:乙酸乙酯=1~1000∶1(体积比)柱层析后,得到黄色固体1.22g,产率为90%。
[0042]1h nmr(600mhz,cdcl3)δ4.33
–
4.28(q,j=6.6hz,2h),3.38(ddd,j=17.2,9.9,6.5hz,2h),2.51
–
2.38(m,1h),1.97(ddd,j=32.4,11.4,5.7hz,1h),1.89(dd,j=11.2,8.0hz,1h),1.87
–
1.81(m,1h),1.81
–
1.77(m,1h),1.76
–
1.74(m,1h),1.73
–
1.67(m,1h),1.67
–
1.59(m,2h),1.59
–
1.49(m,1h),1.48
–
1.38(m,2h),1.37
–
1.34(t,j=6.6hz,3h),1.32
–
1.21(m,1h),1.19(s,2h),1.08
–
1.05(m,2h),1.04
–
0.99(m,4h),0.97
–
0.90(m,1h).
13
c nmr(151mhz,cdcl3)δ164.60(t,j=33.2hz),117.17(t,j=253.6hz),62.80,53.44(t,j=20.9hz),48.88,46.72,40.75,39.90,39.55,37.94,36.26,30.53,28.22,24.25,20.41,17.98,16.55,13.96.ir(kbr)
max
2938,1624,1601,1424,1347,1275,1243,1165,1046,954,735,663cm-1
;ms(ei)m/z 454.1[m]
+
;hrms(ei)m/z[m]
+
calcd for c
19
h
29
o2f2i,454.1175;
found,454.1167.
[0043]
实施例3 β-石竹烯衍生的二氟乙酸乙酯及碘取代的三环化合物3c
[0044]
(1-(4-bromophenyl)-2,2-difluoro-3-((2ar,7bs)-5-iodo-2,2,4a-trimethyl decahydro-7ah-cyclobuta[e]inden-7a-yl)propan-1-one)的制备:
[0045][0046]
向一个干燥无氧氮气保护的三口瓶中依次加入无水碳酸氢钾(0.6g,6mmol),2-苯酚(6mg,0.06mmol),1,4-二氧六环30ml,β-石竹烯(610mg,3mmol),2c(2.16g,6mmol)。随后50℃反应瓶被置于蓝光照射下进行充分搅拌。通过薄层色谱法确认反应终点后,将反应瓶冷却至室温,加入适量水溶液进行猝灭反应,再加入乙酸乙酯萃取水相,合并有机相,有机相用饱和nacl水溶液洗涤,无水硫酸钠干燥,过滤,旋转蒸发回收溶剂,残留物用石油醚:乙酸乙酯=1~100∶1(体积比)柱层析后,得到黄色固体1.56g,产率为92%。
[0047]1h nmr(600mhz,cdcl3)δ7.92(d,j=8.4hz,2h),7.64(d,j=8.6hz,2h),3.43(d,j=9.9hz,1h),3.18(d,j=9.9hz,1h),2.71(ddd,j=29.3,19.8,9.4hz,1h),2.03
–
1.94(m,2h),1.91
–
1.78(m,2h),1.72(ddd,j=23.1,18.0,11.8hz,1h),1.62(ddd,j=16.3,11.7,5.9hz,3h),1.50(t,j=10.4hz,1h),1.32
–
1.29(m,1h),1.25(s,3h),1.07(s,3h),1.02(s,3h).
13
c nmr(151mhz,cdcl3)δ189.07(t,j=32.3hz),132.08,131.53(t,j=3.4hz),131.19,129.70,120.93(t,j=256.70hz),51.67(t,j=20.5hz),50.18,48.27,46.13,44.04,42.28,40.97,37.96,30.21,29.51,23.19,20.61,17.84.ir(kbr)
max 2948,1625,1601,1427,1337,1271,1233,1145,1040,951,733,660cm-1
;ms(ei)m/z 564.1[m]
+
;hrms(ei)m/z[m]
+
calcd for c
23
h
28
brf2io,564.0366;found,564.0361.
[0048]
实施例4 β-石竹烯衍生的二氟乙酸乙酯及碘取代的三环化合物3d
[0049]
(2,2-difluoro-3-((2ar,7bs)-5-iodo-2,2,4a-trimethyldecahydro-7ah-cycl obuta[e]inden-7a-yl)-1-(p-tolyl)propan-1-one)的制备:
[0050][0051]
向一个干燥无氧氮气保护的三口瓶中依次加入无水碳酸氢钾(0.6g,6mmol),2-苯酚(6mg,0.06mmol),1,4-二氧六环30ml,b-石竹烯(610mg,3mmol),2d(1.78g,6mmol)。随后50℃反应瓶被置于蓝光照射下进行充分搅拌。通过薄层色谱法确认反应终点后,将反应瓶冷却至室温,加入适量水溶液进行猝灭反应,再加入乙酸乙酯萃取水相,合并有机相,有机相用饱和nacl水溶液洗涤,无水硫酸钠干燥,过滤,旋转蒸发回收溶剂,残留物用石油醚:乙酸乙酯=1~100∶1(体积比)柱层析后,得到黄色固体1.41g,产率为94%。
[0052]1h nmr(600mhz,cdcl3)δ7.89(d,j=8.0hz,4h),7.21(d,j=8.0hz,4h),3.38(d,j=9.9hz,2h),3.12(d,j=9.9hz,2h),2.72
–
2.60(m,3h),2.36(s,6h),1.95
–
1.89(m,7h),
1.78(ddd,j=13.3,11.5,6.5hz,7h),1.50(s,5h),1.43(t,j=10.3hz,1h),1.37(d,j=13.1hz,1h),1.26(s,1h),1.18(s,2h),0.99(s,2h),0.95(s,1h).
13
c nmr(151mhz,cdcl3)δ187.33,134.70,130.36,130.34,129.53,120.35(t,j=255.9hz),52.03(t,j=20.9hz),50.31,48.43,46.26,44.15,42.45,41.11,38.08,30.34,29.85,29.63,23.31,21.93
–
21.84,20.75,17.96.ir(kbr)
max 2943,1627,1611,1435,1322,1266,1231,1135,1016,953,733,664cm-1
;ms(ei)m/z 500.1[m]
+
;hrms(ei)m/z[m]
+
calcd for c
24
h31f2io,500.4123;found,500.4126.
[0053]
实施例5 β-石竹烯衍生的二氟乙酸乙酯及碘取代的三环化合物3e
[0054]
(1-(4-chlorophenyl)-2,2-difluoro-3-((2ar,7bs)-5-iodo-2,2,4a-trimethy ldecahydro-7ah-cyclobuta[e]inden-7a-yl)propan-1-one)的制备:
[0055][0056]
向一个干燥无氧氮气保护的三口瓶中依次加入无水碳酸氢钾(0.6g,6mmol),2-苯酚(6mg,0.06mmol),1,4-二氧六环30ml,b-石竹烯(610mg,3mmol),2e(1.90g,6mmol)。随后50℃反应瓶被置于蓝光照射下进行充分搅拌。通过薄层色谱法确认反应终点后,将反应瓶冷却至室温,加入适量水溶液进行猝灭反应,再加入乙酸乙酯萃取水相,合并有机相,有机相用饱和nacl水溶液洗涤,无水硫酸钠干燥,过滤,旋转蒸发回收溶剂,残留物用石油醚:乙酸乙酯=1~100∶1(体积比)柱层析后,得到黄色固体1.45g,产率为93%。
[0057]1h nmr(600mhz,cdcl3)δ8.00(d,j=8.0hz,2h),7.47(d,j=8.0hz,2h),3.43(d,j=9.9hz,1h),3.18(d,j=9.8hz,1h),2.80
–
2.64(m,1h),2.04
–
1.95(m,2h),1.92
–
1.80(m,2h),1.71(dd,j=19.8,10.7hz,1h),1.64
–
1.58(m,2h),1.50(t,j=10.3hz,1h),1.33
–
1.27(m,2h),1.06(s,6h),1.02(s,3h).
13
c nmr(151mhz,cdcl3)δ187.80(t,j=32.2hz),139.82,130.47(t,j=3.4hz),129.72,128.05,119.90(t,j=255.9hz),50.64(t,j=20.5hz),49.14,47.23,45.08,42.99,41.24,39.93,36.92,29.17,28.47,22.72
–
22.62,22.15,19.57,16.80,16.53.ir(kbr)
max
2933,1628,1616,1438,1322,1269,1237,1133,1015,957,731,660cm-1
;ms(ei)m/z 520.0[m]
+
;hrms(ei)m/z[m]
+
calcd for c
23
h
28
clf2io,520.0841;found,520.0837.
[0058]
以上实施案例仅用于说明本专利的技术方案,而非对其限制;尽管参照前述实施案例对本专利进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施案例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本专利各实施案例技术方案的精神和范围。
[0059]
致谢:感谢宜宾市引进高层次人才项目(2019yg02)对此实验的经费支持。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips