
 商标分类
商标分类  商标转让
商标转让 
一种2-氰基-4-氟吡啶的制备方法与流程
 2021-02-02 02:02:51|
2021-02-02 02:02:51| 399|
399| 起点商标网
起点商标网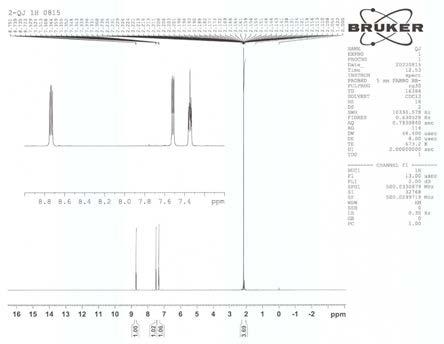
[0001]
本发明涉及一种2-氰基-4-氟吡啶的制备方法。
背景技术:
[0002]
2-氰基-4-氟吡啶是一种新型的含氟医药中间体,在医药领域有广泛的应用。目前2-氰基-4-氟吡啶的合成方法主要是以2-氰基-4-硝基吡啶为原料,经氟代脱硝反应制得。文献(tetrahedron letters,volume 51,issue 14,2010,pages 1906-1909)报道以2-氰基-4-硝基吡啶为原料,分别与氟化钾、四丁基氟化铵、无水四丁基氟化铵在微波促进下进行氟代脱硝反应制备2-氰基-4-氟吡啶,收率分别为57%、小于5%、大于95%。文献(organic letters,volume 7,issue 4,2005,pages 577-579)报道使用市售四丁基氟化铵的四氢呋喃溶液进行氟代脱硝反应制备2-氰基-4-氟吡啶,收率80%。专利(wo2009158011a1,2009)报道使用四丁基氟化铵的四氢呋喃溶液进行氟代脱硝反应制备2-氰基-4-氟吡啶,收率68%。四丁基氟化铵的含水量对氟代脱硝反应影响极大,完全不含水的四丁基氟化铵制备工艺非常复杂(j.am.chem.soc.2005,127,7,2050-2051)。氟化剂四丁基氟化铵分子量较大,作为氟代试剂使用的话,用量非常大,无法工业化生产。另外,经市场调研原料2-氰基-4-硝基吡啶价格非常昂贵,市售产品稀少。查阅文献发现2-氰基-4-硝基吡啶的合成主要是通过4-硝基-n-氧化物与氰化钾或三甲基氰硅烷反应制备得到,收率35-60%(journal of the american chemical society,volume 134,issue 47,2012,pages 19366-19369;us 20020198383a1)。进一步查阅文献发现4-硝基吡啶-n-氧化物主要是以吡啶为原料经氧化、混酸硝化制得(化学试剂,1998,20(4), 240~241)。由于2-氰基-4-硝基吡啶的制备涉及危险的硝化反应;使用剧毒的氰化物;收率低。所以导致2-氰基-4-硝基吡啶的价格极其昂贵。因此,以2-氰基-4-硝基吡啶为原料制备2-氰基-4-氟吡啶路线的原材料价格昂贵、无水四丁基氟化铵的制备工艺复杂,作为氟代剂用量大,该路线无法实现工业化生产。
技术实现要素:
[0003]
本发明的目的是提供一种原料易得、反应条件温和,操作简便、收率高,产品质量好,适合工业化生产的2-氰基-4-氟吡啶制备方法。为了实现发明目的,本发明所提供的2-氰基-4-氟吡啶的制备方法,方法包括以下步骤:(1)以4-氯吡啶-2-甲酸甲酯、氨水为原料,经氨解反应制得4-氯吡啶-2-甲酰胺;(2)在脱水剂的作用下,将4-氯吡啶-2-甲酰胺脱水得到2-氰基-4-氯吡啶;(3)2-氰基-4-氯吡啶经卤素交换反应得到2-氰基-4-氟吡啶。
[0004]
所述步骤(1)的具体反应方式为:将4-氯吡啶-2-甲酸甲酯溶于有机溶剂a,在温度10~50℃和搅拌条件下滴加氨水,滴加完毕继续在温度10~50℃和搅拌条件下反应3~8小时,反应完毕后固液分离得4-氯吡啶-2-甲酰胺。氨水的滴加时间一般为30分钟。
[0005]
上述固液分离前最好先除去有机溶剂a,然后再进行固液分离。
[0006]
所述氨水的浓度为25-28%;所述有机溶剂a选自甲醇、乙醇、异丙醇、四氢呋喃和二
氧六环中的任意一种或二种以上的混合物;所述4-氯吡啶-2-甲酸甲酯与nh3•
h2o的摩尔比为1:1.0~2.5。有机溶剂a的用量以将4-氯吡啶-2-甲酸甲酯完全溶解为底限。
[0007]
所述步骤(2)中,脱水剂为三氯氧磷、三氟乙酸酐和二氯亚砜中的任意一种;当脱水剂选用三氯氧磷或二氯亚砜时:将4-氯吡啶-2-甲酰胺和脱水剂混合,然后让4-氯吡啶-2-甲酰胺和脱水剂进行反应3~12小时,反应完毕,将反应后产物倒入水中进行搅拌析晶,析晶结束进行固液分离得到2-氰基-4-氯吡啶,4-氯吡啶-2-甲酰胺和三氯氧磷反应的温度条件为90~100℃,4-氯吡啶-2-甲酰胺和二氯亚砜的温度条件为70~80℃,将反应后产物倒入水中进行搅拌析晶前最好先脱除过量的脱水剂;当脱水剂选用三氟乙酸酐时:先将4-氯吡啶-2-甲酰胺溶于四氢呋喃、二氧六环中的任意一种溶剂或二种的混合溶剂中,在温度-10~0℃下,滴加三氟乙酸酐(滴加时间一般为30分钟),滴毕,-10~0℃温度下保温反应1~1.5小时,然后升温至30~40℃继续反应2~4小时;反应完毕,除去溶剂,然后将反应后产物倒入水中进行搅拌析晶,析晶结束进行固液分离得到2-氰基-4-氯吡啶。
[0008]
所述4-氯吡啶-2-甲酰胺与脱水剂的摩尔比为比为1:1.2~4.0。
[0009]
所述步骤(3)中卤素交换反应具体方式为:将2-氰基-4-氯吡啶、氟盐和催化剂加入有机溶剂b中,温度110~150℃下反应2~5小时,反应完毕后将反应液加入水中,然后加入有机萃取剂,进行搅拌萃取,萃取完毕后收集有机层,除去有机层中的有机萃取剂就得到2-氰基-4-氟吡啶。所述有机萃取剂为乙酸乙酯。
[0010]
所述氟盐为氟化钾、氟化钠、氟化钙和氟化铯中的任意一种或二种以上的混合物;有机溶剂b为二甲基亚砜、二甲基砜、n,n'-二甲基甲酰胺、n,n'-二甲基乙酰胺、n-甲基吡咯烷酮和环丁砜中的任意一种或二种以上的混合物;催化剂为四丁基溴化铵、四丁基氯化铵、四苯基溴化鏻和四乙基溴化铵中的任意一种或二种以上的混合物;2-氰基-4-氯吡啶与氟盐的摩尔比为1:1.1~3.0;2-氰基-4-氯吡啶与催化剂的摩尔比为1:0.05~0.2。有机溶剂b的用量以将2-氰基-4-氯吡啶完全溶解为底限。
[0011]
除去有机层中的有机萃取剂后,最好再将萃取物加入1,2-二氯乙烷中进行重结晶,重结晶结束后进行固液分离得到2-氰基-4-氟吡啶。
[0012]
本发明以4-氯吡啶-2-甲酸甲酯为原料、经酯的氨解反应、酰胺脱水反应、卤素交换反应制备2-氰基-4-氟吡啶。本发明制备的2-氰基-4-氟吡啶的结构式如下:;其合成路线如下所示:。
[0013]
现有技术的2-氰基-4-氟吡啶的合成主要是以2-氰基-4-硝基吡啶为原料与四丁基氟化铵进行氟代脱硝反应制备2-氰基-4-氟吡啶,存在原料2-氰基-4-硝基吡啶价格昂贵、四丁基氟化铵用量大,氟代脱硝反应受四丁基氟化铵水含量影响大、不适合工业化生产
等问题。本发明使用4-氯吡啶-2-甲酸甲酯为原料经酯的氨解反应得到4-氯吡啶-2-甲酰胺,然后在脱水剂的作用下进行酰胺脱水反应得到2-氰基-4-氯吡啶,最后与氟盐进行卤素交换反应得到的2-氰基-4-氟吡啶,该方法原料易得,工艺简单、收率高,产品纯度高,适合大规模工业化生产。
附图说明
[0014]
图1为实施例1制备的产品的1h-nmr图。
具体实施方式
[0015]
实施例12-氰基-4-氟吡啶的制备方法包括以下步骤:(1)在2l三口瓶安装温度计及套管、恒压滴液漏斗和机械搅拌,向三口瓶中依次加入137.3g(0.8mol)4-氯吡啶-2-甲酸甲酯,823g甲醇,搅拌溶解4-氯吡啶-2-甲酸甲酯,将140.2g(1.0mol)氨水(质量浓度25%)置于恒压滴液漏斗中,温度15~30℃下缓慢滴入上述三口反应瓶中,滴加时间20-30分钟,滴加完毕后继续在15~30℃下搅拌反应5~8小时;反应结束后减压蒸馏除去甲醇,再加入240g水搅拌析晶,抽滤得到4-氯吡啶-2-甲酰胺,收率92.6%,gc纯度99.1%;质谱分析:ms(ei) m/z: 156.0[m
+
]。
[0016]
(2)在1l三口瓶安装温度计及套管、球形冷凝管和机械搅拌,向三口瓶中加入78.3g(0.5mol)4-氯吡啶-2-甲酰胺,470g四氢呋喃,溶清后置于低温浴槽中,降温至-10~0℃,滴加126.0g(0.60mol)三氟乙酸酐,滴加时间20-30分钟,滴毕,-10~0℃保温反应1~1.5小时,然后升温至30~40℃继续反应2~4小时。反应完毕,除去四氢呋喃,加入260g水搅拌析晶,抽滤得到2-氰基-4-氯吡啶,收率87.6%,gc纯度98.3%;质谱分析:ms(ei) m/z: 138.0[m
+
]。
[0017]
(3)在1l三口瓶安装n2侧管、温度计及套管、球形冷凝管、液封、机械搅拌、干燥管;向三口瓶中依次加入69.3g(0.50mol)2-氰基-4-氯吡啶,16.1g(0.05mol)四丁基溴化铵,46.5g(0.80mol)干燥氟化钾,350g n,n'-二甲基甲酰胺,氮气保护,机械搅拌,升温至130~150℃保温反应2~3h。反应完毕将反应液降至室温后倒入680g水中,加入420g乙酸乙酯进行萃取,收集有机层,将有机层中的乙酸乙酯脱除,将萃取物加入1,2-二氯乙烷进行重结晶,重结晶结束后进行固液分离得到2-氰基-4-氟吡啶,收率81.3%,gc纯度99.5%;质谱分析:ms(ei) m/z: 122.0[m
+
]。
[0018]
实施例22-氰基-4-氟吡啶的制备方法包括以下步骤:(1)在2l三口瓶安装温度计及套管、恒压滴液漏斗和机械搅拌,向三口瓶中依次加入137.3g(0.8mol)4-氯吡啶-2-甲酸甲酯、823g四氢呋喃,搅拌溶解4-氯吡啶-2-甲酸甲酯,将150.2g(1.2mol)氨水(质量浓度28%)置于恒压滴液漏斗中,温度35~50℃下缓慢滴入上述三口反应瓶中,滴加时间20-30分钟,滴加完毕继续在温度35~50℃下搅拌反应3~5小时,反应结束后减压蒸馏除去四氢呋喃,然后加入240g水搅拌析晶,抽滤得到4-氯吡啶-2-甲酰胺,收率91.3%,gc纯度99.2%。
[0019]
(2)在1l三口瓶安装温度计及套管、球形冷凝管和机械搅拌,向三口瓶中加入
78.3g(0.5mol)4-氯吡啶-2-甲酰胺、306.6g(2.0mol)三氯氧磷,升温至90~100℃,搅拌反应3~8小时;反应完毕,减压蒸馏除去过量三氯氧磷,然后将反应产物倒入200g冰水混合物中,搅拌析晶,抽滤得到2-氰基-4-氯吡啶,收率88.3%,gc纯度98.6%。
[0020]
(3)在1l三口瓶安装n2侧管、温度计及套管、球形冷凝管、液封、机械搅拌和干燥管;向三口瓶中依次加入69.3g(0.50mol)2-氰基-4-氯吡啶、12.6g(0.03mol)四苯基溴化鏻、54.6g(1.30mol)干燥氟化钠、350g二甲基亚砜,氮气保护,机械搅拌,升温至120~140℃保温反应3~5小时。反应完毕,将反应液降至室温后倒入680g水中,然后加入420g乙酸乙酯进行萃取,萃取完毕收集有机层,减压蒸馏除去有机层中的乙酸乙酯,将萃取物加入1,2-二氯乙烷进行重结晶,重结晶完毕进行固液分离得到2-氰基-4-氟吡啶,收率80.6%,gc纯度99.6%。
[0021]
实施例32-氰基-4-氟吡啶的制备方法包括以下步骤:(1)在2l三口瓶安装温度计及套管、恒压滴液漏斗和机械搅拌,向三口瓶中依次加入137.3g(0.8mol)4-氯吡啶-2-甲酸甲酯、400g乙醇和423g二氧六环,搅拌溶解4-氯吡啶-2-甲酸甲酯;将250.4g(2.0mol)氨水(质量浓度28%)置于恒压滴液漏斗中,温度30~45℃下缓慢滴入上述三口反应瓶中,滴加时间20-30分钟,滴加完毕,在30~45℃下搅拌反应3~4小时,反应结束,减压蒸馏除去乙醇和二氧六环,然后加入240g水搅拌析晶,抽滤得到4-氯吡啶-2-甲酰胺,收率91.7%,gc纯度99.2%。
[0022]
(2)在1l三口瓶安装温度计及套管,球形冷凝管、机械搅拌,向1l三口瓶中加入78.3g(0.5mol)4-氯吡啶-2-甲酰胺,212g(1.8mol)二氯亚砜,升温至70~80℃,反应8~12小时,反应完毕,减压蒸馏除去过量二氯亚砜,然后将反应倒入200g冰水混合物中,搅拌析晶,抽滤得到2-氰基-4-氯吡啶,收率88.8%,gc纯度98.5%。
[0023]
(3)在1l三口瓶安装n2侧管、温度计及套管、球形冷凝管、液封、机械搅拌、干燥管;向三口瓶中依次加入69.3g(0.50mol)2-氰基-4-氯吡啶、12.6g(0.03mol)四苯基溴化鏻、12.6g(0.06mol)四乙基溴化铵、91.1g(0.60mol)干燥氟化铯、150g二甲基砜、200g n-甲基吡咯烷酮,氮气保护,机械搅拌,升温至110~130℃保温反应2~4小时;反应完毕将反应液降至室温后倒入680g水中,然后加入420g乙酸乙酯进行萃取,萃取完毕收集有机层,减压蒸馏除去有机层中的乙酸乙酯,将萃取物加入1,2-二氯乙烷进行重结晶,重结晶完毕进行固液分离得到2-氰基-4-氟吡啶,收率81.2%,gc纯度99.7%。
[0024]
实施例42-氰基-4-氟吡啶的制备方法包括以下步骤:(1)在2l三口瓶安装温度计及套管、恒压滴液漏斗和机械搅拌,向三口瓶中依次加入137.3g(0.8mol)4-氯吡啶-2-甲酸甲酯,823g异丙醇,搅拌溶解4-氯吡啶-2-甲酸甲酯,将112.2g(0.8mol)氨水(质量浓度25%)置于恒压滴液漏斗中,温度10~30℃下缓慢滴入上述三口反应瓶中,滴加时间20-30分钟,滴加完毕后继续在10~30℃下搅拌反应3~6小时;反应结束后减压蒸馏除去甲醇,再加入240g水搅拌析晶,抽滤得到4-氯吡啶-2-甲酰胺,收率92.3%,gc纯度99.4%;质谱分析:ms(ei) m/z: 156.0[m
+
]。
[0025]
(2)在1l三口瓶安装温度计及套管、球形冷凝管和机械搅拌,向三口瓶中加入78.3g(0.5mol)4-氯吡啶-2-甲酰胺,470g二氧六环,溶清后置于低温浴槽中,降温至-10~0
℃,滴加210.0g(1.00mol)三氟乙酸酐,滴加时间20-30分钟,滴毕,-10~0℃保温反应1~1.5小时,然后升温至30~40℃继续反应2~4小时。反应完毕,除去四氢呋喃,加入260g水搅拌析晶,抽滤得到2-氰基-4-氯吡啶,收率87.7%,gc纯度98.5%;质谱分析:ms(ei) m/z: 138.0[m
+
]。
[0026]
(3)在1l三口瓶安装n2侧管、温度计及套管、球形冷凝管、液封、机械搅拌、干燥管;向三口瓶中依次加入69.3g(0.50mol)2-氰基-4-氯吡啶,27.8g(0.10mol)四丁基氯化铵,117.2g(1.50mol)干燥氟化钙,390g环丁砜,氮气保护,机械搅拌,升温至130~140℃保温反应3~4h。反应完毕将反应液降至室温后倒入760g水中,加入440g乙酸乙酯进行萃取,收集有机层,将有机层中的乙酸乙酯脱除,将萃取物加入1,2-二氯乙烷进行重结晶,重结晶结束后进行固液分离得到2-氰基-4-氟吡啶,收率80.8%,gc纯度99.6%;质谱分析:ms(ei) m/z: 122.0[m
+
]。
[0027]
图1是实施例1制备的产品的1h nmr图,其表征的内容如下:1h nmr (cdcl3, 500 mhz),δ: 8.72~8.75 (dd, 1h),7.51~7.53 (dd, 1h),7.33~7.36 (m, 1h)。
[0028]
根据质谱和核磁氢谱分析,实施例1的产品为2-氰基-4-氟吡啶,其结构式如下:。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips