
 商标分类
商标分类  商标转让
商标转让 
一种N-二氟甲基氮杂吲哚类化合物及其合成方法与流程
 2021-02-02 01:02:09|
2021-02-02 01:02:09| 334|
334| 起点商标网
起点商标网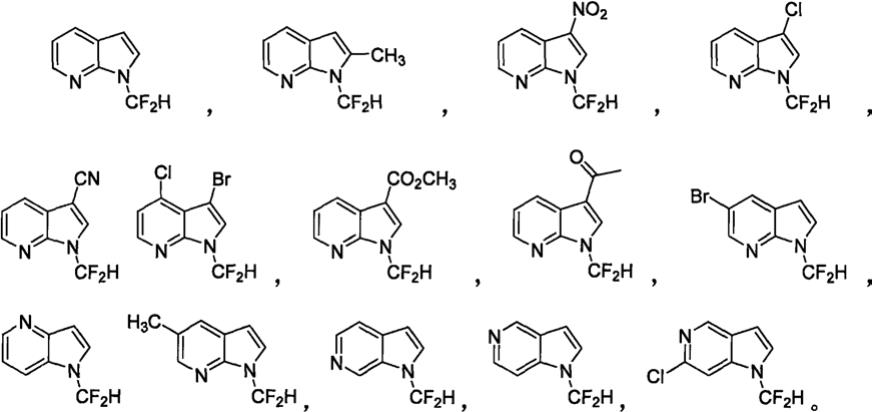
一种n-二氟甲基氮杂吲哚类化合物及其合成方法
技术领域
[0001]
本发明涉及医药化工合成技术领域,特别涉及一种n-二氟甲基氮杂吲哚类化合物及其合成方法。
背景技术:
[0002]
氟原子具有最强的电负性和与氢原子一般大小的原子半径,引入含氟基团,取代氢原子,通常会改变原有分子的物理、化学及生物性质,从而进行药物分子结构的微调和修饰,阻断易代谢位点从而改变药物代谢的途径及代谢速度;并通过分子间氢键的作用,延长药物在体内的作用时间,提高药物的生物利用度和选择(张霁,金传飞,张英俊,chin.j.org.chem.2014,34,662-680)。
[0003]
近年来,二氟甲基(cf2h)因其特殊的化学性质已经引起人们的广泛关注,cf2h和cf3均具有强的亲脂性和吸电子性,cf2h中的氢原子也可以作为氢键的给体参与氢键作用,可以更加有效地增强有机分子的生理活性。因此,对生物活性分子二氟甲基化已成为改造其生物活性的一种有效手段,在新型药物设计和农药开发中日益受到研究人员的重视,如含n-二氟甲基类化合物可作为sdh(琥珀酸脱氢酶)抑制剂类杀菌剂。(陶雪芬,章颖,郑杰锋等,中国医药导报,2019,16(11):38-41)。
[0004]
常见的二氟甲基化试剂有hcf2cl,brcf2co2et,brcf2p(o)(oet)2,tmscf2br,p hscf2h,clcf2coona,ph3p
+
cf2co
2-,tmscf2h,fso2cf2co2ch3等。有关n-二氟甲基化的工作主要有如下一些报道。
[0005]
2007年,胡金波课题组发现了一种以氯二氟甲基苯砜为二氟甲基化试剂,氢氧化钾作为碱,在乙腈溶液中,50℃条件下对仲胺进行二氟甲基化的方法,但是此方法所用的氯二氟甲基苯砜价格比较高,不适合用于大量生产(zheng j.,li y.,hu j.b.,et al.chem.commun.,2007,5149-5151)。
[0006][0007]
2013年,肖吉昌课题组发现了一种以(三苯基磷鎓基)二氟乙酸内盐为二氟甲基化试剂,以对二甲苯为溶剂,90℃条件下生成含n-二氟甲基类化合物的方法。但是此方法存在着所用的二氟甲基化试剂过于昂贵的不足(zheng j.,lin j.h.,chemistry-aeuropean journal,2013,19(45):15261-15266)。
[0008]
[0009]
2014年,prakash报道了一种在碘化锂与(三氟甲基)三甲基硅烷共同作用下,以三甘醇二甲醚为溶剂,170℃条件下对仲胺进行二氟甲基化的方法,但是此方法所需的反应温度过高(prakash g.k.s.,krishnamoorthy s.et al.organic lett.,2014,16(1),54-57)。
[0010][0011]
2018年,janaal课题组使用廉价易得且环境友好的brcf2co2et试剂,在lioh的作用下,对具有甲苯磺酰基保护基的芳胺进行了n-二氟甲基化反应(polley a.,bairyet g.,janaal r.,adv.synth.catal.,2018,360,4161-4167)。
[0012][0013]
2018年,李晓飞课题组开发了一种简单有效的咪唑和吡唑的n-二氟甲基化方法。该反应利用brcf2p(o)(oet)2作为二氟卡宾供体,对咪唑和吡唑进行n-二氟甲基化反应(mao t.,zhao l.,li x.f.,et al.tetrahedron lett.,2018,59,2752-2754)。
[0014][0015]
2019年,汤日元课题组报道了亚硫酸盐促进的唑类n-二氟甲基化和硫化反应获得n-二氟甲基硫脲,突破了使用昂贵且不易获得的试剂氟磺酰基二氟乙酸三甲基硅酯并且反应底物范围小的限制,在该反应中,廉价的brcf2co2et和无毒硫元素分别用作二氟甲基化和硫化试剂。各种唑类,包括苯并咪唑类、咪唑类和三唑类,反应表现良好,可获得中等至良好收率的各种唑类硫脲(deng j.c.,gao y.c.,tang r.y.,et al.org.lett.2019,21,545-548)。
[0016][0017]
2019年,江焕峰课题组报道了一种腙类化合物n-二氟甲基化方法,以芳基乙酮对甲苯磺酰腙为底物,溴二氟甲基三甲基硅烷为二氟甲基试剂,碳酸铯为碱,苄基三乙基氯化铵(tebac)为添加剂,以甲苯为溶剂,敞开体系中90℃搅拌反应12小时。反应可获得中等至良好收率的各种腙类化合物,但是此方法所需的二氟甲基化试剂昂贵(huang y.,lin z.,org.chem.front.,2019,6,2462-2466)。
[0018][0019]
氮杂吲哚类化合物,更确切地说是吡咯并吡啶,是一类重要的有机化工产品中间体,也是许多具有生理活性的天然产物及药物的基本骨架。研究发现,具有这类结构的化合物用途十分广泛,可用于医药、染料、农药、香料、发光材料和其它精细化工产品的中间体。(wu q.g,jamesa.l.,tao.y,et al.,inorg.chem.,2000(39):5248-5254;zhao s.b.,wang s.n.,chem.soc.rev.,2010(39):3142-3156;walker s.r.,carter e.j.,huff b.c.,et al.,chem.rev.,2009(109):3080-3098)
[0020]
近年来,由于其特定的物化性质,常在功能材料合成与配位化学中得到较多的应用;更为重要的是,它与吲哚、嘌呤等在结构上的相似性,可作为这些类似化合物的生物电子等排体,可用于抗癌、抗菌、抗病毒、抗抑郁以及治疗高血压等。其已成为杂环类化合物热点研究方向之一,市场需求巨大,发展前景广阔(moore n.a.,tupper d.e.,horen t.m.,drugs future 1994(19):114-117;田海滨,尹端,张春富等,中国药物化学杂志,2002,12(4):214-218)。但是目前还没有利用氮杂吲哚为原料直接合成n-二氟甲基氮杂吲哚类化合物的报道。
技术实现要素:
[0021]
为了解决以上技术问题,本发明的目的在于提供一种n-二氟甲基氮杂吲哚类化合物及其合成方法,具有通用、简便、高效的特点。
[0022]
为了实现上述目的,本发明采用的技术方案是:
[0023]
一种n-二氟甲基氮杂吲哚类化合物,所述的n-二氟甲基氮杂吲哚类化合物结构式为:
[0024][0025]
所述r1包括氢、甲基、烷基、烷氧基、氨基、烷胺基、氰基、硝基、氟、氯、溴、碘、乙酰基、磺酰基、酯基、苯基、芳基基、吡啶基、呋喃基、噻吩基、萘环、芳杂基;
[0026]
r2包括氢、甲基、烷基、烷氧基、氨基、烷胺基、氰基、硝基、氟、氯、溴、碘、乙酰基、磺酰基、酯基、苯基、芳基基、吡啶基、呋喃基、噻吩基、萘环、芳杂基;
[0027]
r3包括氢、甲基、烷基、烷氧基、氨基、烷胺基、氰基、硝基、氟、氯、溴、碘、乙酰基、磺酰基、酯基、苯基、芳基基、吡啶基、呋喃基、噻吩基、萘环、芳杂基;
[0028]
r4包括氢、甲基、烷基、烷氧基、氨基、烷胺基、氰基、硝基、氟、氯、溴、碘、乙酰基、磺
酰基、酯基、苯基、芳基基、吡啶基、呋喃基、噻吩基、萘环、芳杂基;
[0029]
r5包括氢、甲基、烷基、烷氧基、氨基、烷胺基、氰基、硝基、氟、氯、溴、碘、乙酰基、磺酰基、酯基、苯基、芳基基、吡啶基、呋喃基、噻吩基、萘环、芳杂基。
[0030]
所述n-二氟甲基氮杂吲哚类化合物具体为
[0031][0032]
一种n-二氟甲基氮杂吲哚类化合物的合成方法,包括以下步骤;
[0033]
在反应器中,加入氮杂吲哚类化合物、二氟甲基化试剂、碱和有机溶剂,室温搅拌反应6~12小时,产物经分离纯化,得到所述n-二氟甲基氮杂吲哚类化合物。
[0034]
所述二氟甲基化试剂为brcf2co2et,brcf2co2k,clcf2co2na,tmscf2br,brcf2p(o)(oet)2,fso2cf2co2ch3。
[0035]
所述氮杂吲哚类化合物与二氟甲基化试剂的摩尔比为1∶1.2~3。
[0036]
所述氮杂吲哚类化合物与二氟溴乙酸乙酯的最优摩尔比为1∶3。
[0037]
所述碱为叔丁醇钾、叔丁醇锂、碳酸钾、甲醇钠、乙醇钠、碳酸铯、三乙胺、2,6-二甲基吡啶和n,n-二异丙基乙胺中的一种,最优选为叔丁醇钾。
[0038]
所述碱的加入量与二氟溴乙酸乙酯的摩尔比为1∶1。
[0039]
所述的有机溶剂为乙酸乙酯,甲苯、氯苯、乙腈、二氯甲烷、1,2-二氯乙烷、四氢呋喃或1,4-二氧六环,最优选为乙腈。
[0040]
本发明的有益效果:
[0041]
(1)所用的二氟甲基试剂廉价易得,反应条件温和,室温即可完成反应。
[0042]
(2)本发明的合成方法对官能团适应性好、对底物适应性广,各种取代基都可以实现n-二氟甲基化,具有良好的工业应用前景。
[0043]
(3)反应步骤简单,且合成的n-二氟甲基氮杂吲哚是一类新化合物。
附图说明
[0044]
图1为n-二氟甲基-7-氮杂吲哚核磁共振氢谱图。
[0045]
图2为n-二氟甲基-6-氮杂吲哚核磁共振氢谱图。
[0046]
图3为n-二氟甲基-4-氮杂吲哚核磁共振氢谱图。
[0047]
图4为n-二氟甲基-2-甲基-7-氮杂吲哚核磁共振氢谱图。
[0048]
图5为n-二氟甲基-3-硝基-7-氮杂吲哚核磁共振氢谱图。
[0049]
图6为n-二氟甲基-3-氯-7-氮杂吲哚核磁共振氢谱图。
[0050]
图7为n-二氟甲基-3-氰基-7-氮杂吲哚核磁共振氢谱图。
[0051]
图8为n-二氟甲基-3-溴-4-氯-7-氮杂吲哚核磁共振氢谱图。
[0052]
图9为n-二氟甲基-3-甲酸甲酯基-7-氮杂吲哚核磁共振氢谱图。
[0053]
图10为n-二氟甲基-3-乙酰基-7-氮杂吲哚核磁共振氢谱图。
[0054]
图11为n-二氟甲基-5-溴-7-氮杂吲哚核磁共振氢谱图。
[0055]
图12为n-二氟甲基-5-甲基-7-氮杂吲哚核磁共振氢谱图。
[0056]
图13为n-二氟甲基-6-氯-5-氮杂吲哚核磁共振氢谱图。
[0057]
图14为n-二氟甲基-4-氯-6-氮杂吲哚核磁共振氢谱图。
具体实施方式
[0058]
下面结合附图对本发明作进一步详细说明。
[0059]
如图-图14所示,具体为:
[0060]
实施例1
[0061]
向25ml耐压瓶中依次加入7-氮杂吲哚(0.5mmol),二氟溴乙酸乙酯(2.0mmol),叔丁醇钾(2.0mmol),乙腈(2ml),充氮密封耐压瓶,室温搅拌反应12h。向反应液中加入20ml水,再用乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩,得到粗产物。粗产物用石油醚和乙酸乙酯的混合溶剂作洗脱液经过柱层析得到目标化合物,得率为90%。其核磁共振氢谱数据为:
[0062]1h nmr(300mhz,cdcl3)δ8.35(d,j=4.6hz,1h),7.93(d,j=7.9hz,1h),7.89(t,j=60.1hz,1h),7.51(d,j=3.8hz,1h),7.19(dd,j=7.9,4.8hz,1h),6.63(d,j=3.5hz,1h).
[0063]
实施例2
[0064]
向25ml耐压瓶中依次加入6(或4)-氮杂吲哚(0.5mmol),二氟溴乙酸乙酯(2.0mmol),叔丁醇钾(2.0mmol),乙腈(2ml),充氮密封耐压瓶,将其放入预先加热到80℃的油浴中反应12h。反应结束后,冷却至室温。向反应液中加入20ml水,再用乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩,得到粗产物。粗产物用石油醚和乙酸乙酯的混合溶剂作洗脱液经过柱层析得到目标化合物,得率为74~85%。所得产物的核磁共振氢谱数据为:
[0065]1h nmr(300mhz,cdcl3)δ8.99(s,1h),8.39(d,j=5.3hz,1h),7.55(d,j=4.8hz,1h),7.43(d,j=3.6hz,1h),7.34(t,j=60.0hz,1h),6.65(d,j=3.2hz,1h).
[0066]1h nmr(300mhz,cdcl3)δ8.57(d,j=4.7hz,1h),7.90(d,j=8.0hz,1h),7.50(d,j=3.7hz,1h),7.24(t,j=60.1hz,1h),7.25-7.17(m,2h),6.85(d,j=3.7hz,
1h)。
[0067]
实施例3
[0068]
向25ml耐压瓶中依次加入3位(或4位,或5,或6位)取代的7-氮杂吲哚(0.5mmol),二氟溴乙酸乙酯(2.0mmol),叔丁醇钾(2.0mmol),乙腈(2ml),充氮密封耐压瓶,室温搅拌反应12h。向反应液中加入20ml水,再用乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩,得到粗产物。粗产物用石油醚和乙酸乙酯的混合溶剂作洗脱液经过柱层析得到目标化合物,得率为65~89%。所得产物的核磁共振氢谱数据为:
[0069]1h nmr(300mhz,cdcl3)δ8.23(d,j=4.9hz,1h),7.92(t,j=59.3hz,1h),7.77(d,j=7.6hz,1h),7.11(dd,j=7.7,4.8hz,1h),6.29(s,1h),2.62(s,3h).
[0070]1h nmr(300mhz,cdcl3)δ8.95(d,j=8.1hz,1h),8.86(d,j=6.0hz,1h),8.71(s,1h),8.70(t,j=59.5hz,1h),7.65(t,j=7.2hz,1h).
[0071]1h nmr(300mhz,cdcl3)δ8.38(d,j=3.4hz,1h),7.93(d,j=7.9hz,1h),7.87(t,j=59.8hz,1h),7.48(s,1h),7.32-7.19(m,1h).
[0072]1h nmr(300 mhz,cdcl3)δ8.49(d,j=3.4 hz,1h),8.13(d,j=7.7 hz,1h),8.06(s,1h),7.88(t,j=59.5 hz,1h),7.39(dd,j=8.0,4.9 hz,1h).
[0073]1h nmr(300 mhz,cdcl3)δ8.21(d,j=5.2 hz,1h),7.81(t,j=60.1 hz,1h),7.59(s,1h),7.21(d,j=5.3 hz,1h).
[0074]1h nmr(300 mhz,cdcl3)δ8.44(d,j=7.6 hz,1h),8.39(d,j=3.7 hz,1h),8.18(s,1h),7.87(t,j=60.2 hz,1h),7.38-7.24(m,1h),3.94(s,1h).
1
h nmr(300 mhz,cdcl3)δ8.66(d,j=7.9 hz,1h),8.41(d,j=3.6 hz,1h),8.12(s,1h),7.90(t,j=60.0 hz,1h),7.34(dd,j=8.0,4.6 hz,1h),2.59(s,3h).
[0075]1h nmr(300 mhz,cdcl3)δ8.36(d,j=2.0 hz,1h),8.04(d,j=2.0 hz,1h),7.80(t,j=60.3 hz,1h),7.51(d,j=3.9 hz,1h),6.57(d,j=3.6 hz,1h).
[0076]1h nmr(300 mhz,cdcl3)δ8.17(s,1h),7.84(t,j=59.5 hz,1h),7.70(s,1h),7.46(s,1h),6.54(s,1h),2.43(s,3h).
[0077]
实施例4
[0078]
向25ml耐压瓶中依次加入取代的6(或5)-氮杂吲哚(0.5mmol),二氟溴乙酸乙酯(2.0mmol),叔丁醇钾(2.0mmol),乙腈(2ml),充氮密封耐压瓶,充氮密封耐压瓶,将其放入预先加热到80℃的油浴中反应12h。反应结束后,冷却至室温。向反应液中加入20ml水,再用乙酸乙酯萃取,有机相用无水硫酸钠干燥,过滤,滤液浓缩,得到粗产物。粗产物用石油醚和乙酸乙酯的混合溶剂作洗脱液经过柱层析得到目标化合物,得率为50~85%。所得产物的核磁共振氢谱数据为:
[0079]1h nmr(300mhz,cdcl3)δ8.71(s,1h),7.53(s,1h),7.33(d,j=3.6hz,1h),7.28(t,j=59.5hz,1h),6.73(d,j=4.2hz,1h).
[0080]1h nmr(300mhz,cdcl3)δ8.91(s,1h),8.39(s,1h),7.52(d,j=4.5hz,1h),7.36(t,j=60.1hz,1h),6.80(d,j=3.6hz,1h).
[0081]
实施例5
[0082]
反应步骤与实施例1完全相同,不同之处在于:反应溶剂为dmf,n-二氟甲基-7-氮杂吲哚收率为52%;反应溶剂为thf,n-二氟甲基-7-氮杂吲哚收率为62%;反应溶剂为dcm,n-二氟甲基-7-氮杂吲哚收率为84%;反应溶剂为dmso,n-二氟甲基-7-氮杂吲哚收率为0%。
[0083]
实施例6
[0084]
反应步骤与实施例1完全相同,不同之处在于:碱为k2co3,n-二氟甲基-7-氮杂吲哚收率为75%;加入lioh时,n-二氟甲基-7-氮杂吲哚收率为77%;加入cs2co3时,n-二氟甲基-7-氮杂吲哚收率为80%;加入et3n时,n-二氟甲基-7-氮杂吲哚收率为65%。
[0085]
实施例7
[0086]
反应步骤与实施例1完全相同,不同之处在于:反应二氟甲基化试剂为brcf2p(o)(oet)2,n-二氟甲基-7-氮杂吲哚收率为80%。
[0087]
实施例8
[0088]
反应步骤与实施例1完全相同,不同之处在于:反应二氟甲基化试剂为fso2cf2co2ch3,n-二氟甲基-7-氮杂吲哚收率为83%。
[0089]
实施例9
[0090]
反应步骤与实施例1完全相同,不同之处在于:反应二氟甲基化试剂为tmscf2br,n-二氟甲基-7-氮杂吲哚收率为86%。
[0091]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其它的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips