
 商标分类
商标分类  商标转让
商标转让 
一种FGFR4抑制剂、制备方法、药物组合物及其应用与流程
 2021-02-02 01:02:58|
2021-02-02 01:02:58| 298|
298| 起点商标网
起点商标网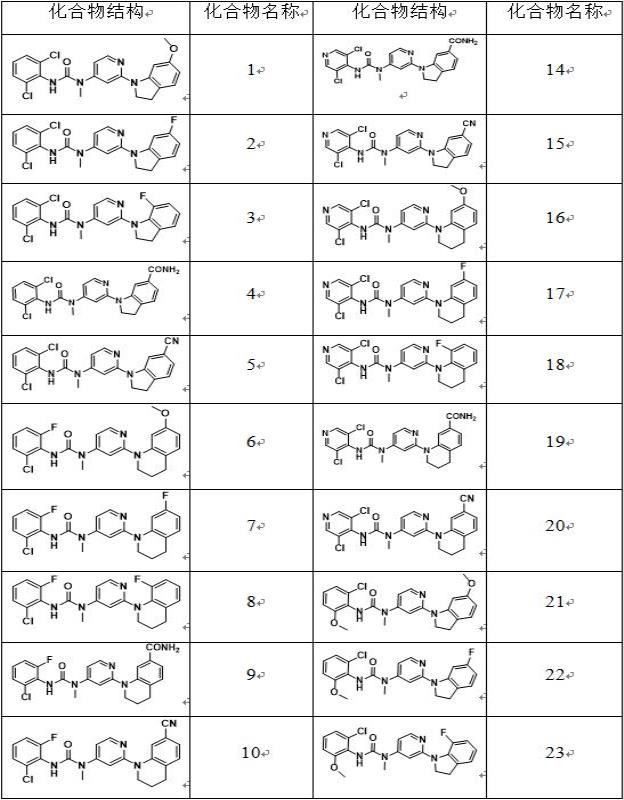
一种fgfr4抑制剂、制备方法、药物组合物及其应用
技术领域
[0001]
本发明属于生物医药技术领域,具体涉及一种fgfr4抑制剂、制备方法、药物组合物及其应用。
背景技术:
[0002]
癌症是威胁人类健康的重大疾病之一,目前癌症的主要治疗方式包括药物治疗、手术治疗和放射治疗等,其中药物治疗是最常用的治疗方式之一。传统的细胞毒药物无法区分肿瘤细胞和正常细胞,常导致严重的副作用,而靶向药物以肿瘤细胞作为特异性靶点,能准确作用于肿瘤,在极大提高癌症的治疗水平的同时能够有效降低不良反应率。
[0003]
fgfr(fibroblast growth factors receptor,成纤维细胞生长因子)属于受体蛋白酪氨酸激酶,该家族主要包括fgfr1、fgfr2、fgfr3、fgfr4。fgfr参与细胞增殖、凋亡、迁移、新生血管生成等多个过程。研究发现,fgfr激活与肝癌、膀胱癌、肺癌、乳腺癌等相关,并且在肿瘤新生血管生成、肿瘤的侵袭与转移等过程中也发挥重要作用,因此,fgfr可以作为肿瘤治疗的重要靶点。
[0004]
fgf(fibroblast growth factor,成纤维生长因子)与受体fgfr结合后使受体胞内酪氨酸残基或靶蛋白酪氨酸残基磷酸化激活,然后通过多种细胞内信号传导分子活化相关传导途径。fgf诱导的下游信号有pkc路径、ras/raf/mek/erk路径、jak/stat路径、piek路径等。fgf信号可以激活蛋白激酶erk1和erk2,且激酶活性持续时间显著长于表皮生长因子(egf)诱导的磷酸化激酶持续时间。不同路径的活化还能磷酸化myc等早期转录因子,促使相关靶基因转录;同时fgfr磷酸化后还可直接转入细胞核内发挥作用。fgfr是肝脏中主要的fgf受体亚型,目前发现的20多种成纤维生长因子中有10个能与fgfr4结合。近年来研究表明,fgfr4的改变,如过表达、突变、异位等与多种癌症的进展有关。因此提高对fgfr4的抑制作用,有助于提高癌症的治疗水平和有效降低不良反应率,从而解决现有抗癌药物的耐药性能,提高药物的靶向作用。
技术实现要素:
[0005]
本发明的研究者发现具有式i结构的化合物对fgfr4具有较好的抑制活性,有助于提高药物对癌症的治疗效果和药物的靶向作用,在制备治疗肿瘤的药物方面具有非常良好的应用前景。
[0006]
该式i结构的化合物为一种fgfr4抑制剂,本发明的目的在于提供一种fgfr4抑制剂、制备方法、药物组合物及其应用,为实现上述发明目的,本发明采用的技术方案如下:为实现上述发明目的,本发明采用的技术方案如下:本发明第一方面,提供一种具有式i结构的化合物或其药学上可接受的盐:
其中,r1代表取代或不取代的苯基或芳杂环基,r2代表h、f、cl、br、meo、cn、conh2,n为1或2。
[0007]
本发明第二方面,提供一种如上所述的具有式i结构的化合物或其药学上可接受的盐的制备方法,其特征在于,包括如下步骤:中间体iv的合成:在第一预设温度下,将具有式ii结构的化合物ii、具有式iii结构的化合物iii以及第一碱溶于第一反应溶剂中进行反应,得到具有式iv结构的中间体iv;化合物i的合成:在第二预设温度下,将所述中间体iv、具有式v结构的化合物v、催化剂、配体以及第二碱溶于第二反应溶剂中进行反应,得到具有式i结构的化合物i。
[0008]
在一个实施例中,所述第一碱选自三乙胺、二异丙基乙胺、n-甲基吗啉、碳酸钾、氢氧化钠、氢氧化锂、氢化钠、叔丁醇钠、叔丁醇钾和六甲基氨基锂中的至少一种;和/或,所述第二碱选自碳酸铯、叔丁醇钠、叔丁醇钾、碳酸钾中的至少一种。
[0009]
在一个实施例中,所述第一反应溶剂为二氯甲烷、乙腈(ch3cn)、四氢呋喃(thf)、二氧六环、甲苯和n,n-二甲基甲酰胺(dmf)中的至少一种;和/或,所述第二反应溶剂为二氧六环、n,n-二甲基甲酰胺和甲苯中的至少一种。
[0010]
在一个实施例中,所述催化剂选自醋酸钯(pd(oac)2)、三(二亚苄基丙酮)二钯(pd2(dba)3)和1,1'-[双(二苯基膦基)二茂铁]二氯化钯(pd(dppf)cl2)中的至少一种;和/或,所述配体选自1,1'-联萘-2,2'-双二苯膦(binap)、2-二环己基膦-2',6'-双甲氧基联苯(s-phos)和4,5-双(二苯基膦)-9,9-二甲基氧杂蒽(xantphos)中的至少一种。
[0011]
在一个实施例中,所述第一预设温度为0℃~80℃;和/或,所述第二预设温度为80℃~120℃。
[0012]
本发明第三方面,提供一种药物组合物,包含如上所述的具有式i结构的化合物或其药学上可接受的盐,以及药用载体或稀释剂。
[0013]
本发明第四方面,提供一种物质在制备治疗肿瘤的药物中的应用,所述物质包括如上所述的具有式i结构的化合物或其药学上可接受的盐;或者,所述物质包括如上所述的具有式i结构的化合物或其药学上可接受的盐作为活性成分的药物组合物;或者,所述物质包括如上所述的药物组合物。
[0014]
在一个实施例中,所述肿瘤选自皮肤癌、膀胱癌、卵巢癌、乳腺癌、胃癌、前列腺癌、
[0016]
本发明所获得的有益效果至少在于:(1)本发明提供一种式i结构的化合物,该化合物具有良好的抑制fgfr4活性的能力,可以作为有效的fgfr4抑制剂,有助于提高癌症的治疗水平和有效降低不良反应率。
[0017]
(2)本发明提供的药物组合物具有良好的多种抗肿瘤药理活性,有助于提高药物对癌症的治疗效果和药物的靶向作用。
具体实施方式
[0018]
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
[0019]
本发明实施例中的一些定义如下:“药学上可接受的盐”是指那些保留母体化合物的生物有效性及特性的盐。该盐包括:酸加成盐,其是通过母体化合物的游离碱与无机酸或与有机酸的反应而获得的;所述无机酸诸如盐酸、氢溴酸、氢碘酸、硝酸、磷酸、硫酸及高氯酸等;所述有机酸诸如乙酸、草酸、(d)或(l)苹果酸、马来酸、甲烷磺酸、乙烷磺酸、对甲苯磺酸、水杨酸、酒石酸、苯磺酸(苯磺酸盐)、苯甲酸、樟脑磺酸、柠檬酸、富马酸、葡萄糖酸、谷氨酸、羟乙磺酸、乳酸、马来酸、扁桃酸、黏液酸、双羟萘酸、泛酸、琥珀酸、酒石酸或丙二酸等;优选为盐酸或(l)-苹果酸;或者当母体化合物中存在的酸质子被置换为金属离子或与有机碱配位时,形成盐,所述金属离子例如碱金属离子、碱土离子或铝离子;所述有机碱诸如乙醇胺、二乙醇胺、三乙醇胺、缓血酸胺、n-甲基葡糖胺及类似物。
[0020]“药物组合物”是指一种或多种本文中所述的化合物或其生理学上可接受的盐与其它化学成分(诸如生理学上可接受的载体及赋形剂)的混合物。药物组合物的目的旨在促进化合物给予生物。
[0021]“载体”当用于本文中时是指对生物不产生明刺激且不会消除所给予的化合物的生物活性及特性的载体或稀释剂。
[0022]“苯基”是指以苯环为官能团的基团。
[0023]“芳基”是指具有完全共轭的π电子系统的全碳单环或稠环多环(亦即,共享相邻碳原子对的环)基团。优选地,芳基在环中具有6到12个碳原子。
[0024]
本发明实施例提供的具有式i结构的化合物可具有一个或多个不对称中心;该化合物因此可以个别(r)-立体异构体或(s)-立体异构体形式制备或以其混合物形式制备。除
非另有说明,否则本说明书及权利要求中的特定化合物的描述或名称意欲包括个别对映异构体与其外消旋混合物或其它混合物。用于测定立体化学构型及分离立体异构体的方法在本领域中是熟知的(参见"advanced organic chemistry"的第4章中的论述,第4版,j. march,johnwiley及sons,new york,1992)。因此,本发明亦涵盖具有抑制fgfr4活性的任何立体异构形式、其相应对映异构体(d-异构体及l-异构体或(+)异构体及(-)异构体)及其非对映异构体及其混合物且不限于任一种立体异构形式。
[0025]
实施例13-(2,6-二氯苯基)-1-(2-(6-甲氧基吲哚啉-1-基)吡啶-4-基)-1-甲基脲化合物1c的合成:将化合物1a(37.4g,200.0mmol)、1b(37.2g,200.0mmol)、碳酸钾(41.4g,300.0mmol)溶于乙腈(500ml)中,室温反应10小时,tlc监测反应,反应完毕后过滤,滤液浓缩、柱层析分离得到类白色(化合物1c)58.3g,收率为78.3%。
[0026]
化合物1的合成:将化合物1c(3.7g,10.0mmol)、化合物1d(1.5g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到白色固体(化合物1)2.3g,收率为52.0%。esi(+) m/z=443.1。
[0027]
实施例23-(2,6-二氯苯基)-1-(2-(6-氟吲哚啉-1-基)吡啶-4-基)-1-甲基脲按照实施例1进行化合物1c的合成。
[0028]
化合物2的合成:将化合物1c(3.7g,10.0mmol)、化合2a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到淡黄色固体(化合物2)2.7g,收率为62.8%。esi(+) m/z=431.1。
[0029]
实施例33-(2,6-二氯苯基)-1-(2-(7-氟吲哚啉-1-基)吡啶-4-基)-1-甲基脲
按照实施例1进行化合物1c的合成。
[0030]
化合物3的合成:将化合物1c(3.7g,10.0mmol)、化合3a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到淡黄色固体(化合物3)2.0g,收率为46.5%。esi(+) m/z=431.1。
[0031]
实施例41-(4-(3-(2,6-二氯苯基)-1-甲基脲基)吡啶-2-基)吲哚啉-6-甲酰胺按照实施例1进行化合物1c的合成。
[0032]
化合物4的合成:将化合物1c(3.7g,10.0mmol)、化合4a(1.6g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到淡黄色固体(化合物4)3.1g,收率为68.1%。esi(+) m/z=456.1。
[0033]
实施例51-(2-(6-氰基吲哚啉-1-基)吡啶-4-基)-3-(2,6-二氯苯基)-1-甲基脲按照实施例1进行化合物1c的合成。
[0034]
化合物5的合成:将化合物1c(3.7g,10.0mmol)、化合5a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到淡黄色固体(化合物5)2.5g,收率为57.2%。esi(+) m/z=438.1。
[0035]
实施例63-(2-氯-6-氟苯基)-1-(2-(7-甲氧基-3,4-二氢喹啉-1(2h)-基)吡啶-4-基)-1-甲基脲
化合物6b的合成:将化合物6a(17.1g,100.0mmol)、1b(18.6g,100.0mmol)、碳酸钾(20.7g,150.0mmol)溶于乙腈(300ml)中,室温反应10小时,tlc监测反应,反应完毕后过滤,滤液浓缩、柱层析分离得到类白色(化合物6b)25.4g,收率为71.1%。
[0036]
化合物6的合成:将化合物6b(3.6g,10.0mmol)、化合物6d(1.6g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物6)2.9g,收率为65.6%,esi(+) m/z=441.1。
[0037]
实施例73-((2-氯-6-氟苯基)-1-(2-(7-氟-3,4-二氢喹啉-1(2h)-基)吡啶-4-基)-1-甲基脲按照实施例6进行化合物6b的合成。
[0038]
化合物7的合成:将化合物6b(3.6g,10.0mmol)、化合物7a(1.5g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物7)2.4g,收率为54.1%,esi(+) m/z=445.1。
[0039]
实施例83-(2-氯-6-氟苯基)-1-(2-(8-氟-3,4-二氢喹啉-1(2h)
-ꢀ
基)吡啶-4-基)-1-甲基脲按照实施例6进行化合物6b的合成。
[0040]
化合物8的合成:将化合物6b(3.6g,10.0mmol)、化合物8a(1.5g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物8)3.1g,收率为69.8%,esi(+) m/z=445.1。
[0041]
实施例91-(4-(2-氯-6-氟苯基)-1-甲基脲基)吡啶-2-基)-1,2,3,4-四氢喹啉-7-甲酰胺按照实施例6进行化合物6b的合成。
[0042]
化合物9的合成:将化合物6b(3.6g,10.0mmol)、化合物9a(1.8g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物9)2.7g,收率为57.4%,esi(+) m/z=470.1。
[0043]
实施例101-(2-(7-氰基-3,4-二氢喹啉-1(2h)-基)吡啶-4-基)-3-(2-氯-6-氟苯基)-1-甲基脲按照实施例6进行化合物6b的合成。
[0044]
化合物10的合成:将化合物6b(3.6g,10.0mmol)、化合物10a(1.6g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物10)3.0g,收率为66.7%,esi(+) m/z=452.1。
[0045]
实施例113-(3,5-二氯吡啶-4-基)-1-(2-(6-甲氧基吲哚啉-1-基)吡啶-4-基)-1-甲基脲化合物11b的合成:将化合物11a(18.8g,100.0mmol)、1b(18.6g,100.0mmol)、碳酸钾(20.7g,150.0mmol)溶于乙腈(300ml)中,室温反应10小时,tlc监测反应,反应完毕后过滤,滤液浓缩、柱层析分离得到类白色(化合物11b)27.1g,收率为72.7%。
[0046]
化合物11的合成:将化合物11b(3.7g,10.0mmol)、化合物1d(1.5g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机
层浓缩,柱层析分离得到类白色固体(化合物11)2.0g,收率为45.1%,esi(+) m/z=444.1。
[0047]
实施例123-(3,5-二氯吡啶-4-基)-1-(2-(6-氟吲哚啉-1-基)吡啶-4-基)-1-甲基脲按照实施例11进行化合物11b的合成。
[0048]
化合物12的合成:将化合物11b(3.7g,10.0mmol)、化合2a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物12)2.6g,收率为60.3%,esi(+) m/z=432.1。
[0049]
实施例133-(3,5-二氯吡啶-4-基)-1-(2-(7-氟吲哚啉-1-基)吡啶-4-基)-1-甲基脲按照实施例11进行化合物11b的合成。
[0050]
化合物13的合成:将化合物11b(3.7g,10.0mmol)、化合3a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物13)3.1g,收率为71.9%,esi(+) m/z=432.1。
[0051]
实施例141-(4-(3-(3,5-二氯吡啶-4-基)-1-甲基脲基)吡啶-2-基)吲哚啉-6-甲酰胺按照实施例11进行化合物11b的合成。
[0052]
化合物14的合成:将化合物11b(3.7g,10.0mmol)、化合4a(1.6g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物14)2.5g,收率为54.8%,esi(+) m/z=457.1。
[0053]
实施例151-(2-(6-氰基吲哚啉-1-基)吡啶-4-基)-3-(3,5-二氯吡啶-4-基)-1-甲基脲按照实施例11进行化合物11b的合成。
[0054]
化合物15的合成:将化合物11b(3.7g,10.0mmol)、化合5a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物15)2.8g,收率为63.9%,esi(+) m/z=439.1。
[0055]
实施例163-(3-氯-5-氟吡啶-4-基)-1-(2-(7-甲氧基-3,4-二氢喹啉-1(2h)-基)吡啶-4-基)-1-甲基脲化合物16b的合成:将化合物16a(17.2g,100.0mmol)、1b(18.6g,100.0mmol)、碳酸钾(20.7g,150.0mmol)溶于乙腈(300ml)中,室温反应10小时,tlc监测反应,反应完毕后过滤,滤液浓缩、柱层析分离得到类白色(化合物16b)24.5g,收率为68.4%。
[0056]
化合物16的合成:将化合物16b(3.6g,10.0mmol)、化合物6d(1.6g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物16)2.2g,收率为49.9%,esi(+) m/z=442.1。
[0057]
实施例173-(3-氯-5-氟吡啶-4-基)-1-(2-(7-氟-3,4-二氢喹啉-1(2h)
-ꢀ
基)吡啶-4-基)-1-甲基脲按照实施例16进行化合物16b的合成。
[0058]
化合物17的合成:
将化合物16b(3.6g,10.0mmol)、化合物7a(1.5g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物17)2.9g,收率为65.2%,esi(+) m/z=446.1。
[0059]
实施例183-(3-氯-5-氟吡啶-4-基)-1-(2-(8-氟-3,4-二氢喹啉-1(2h)
-ꢀ
基)吡啶-4-基)-1-甲基脲按照实施例16进行化合物16b的合成。
[0060]
化合物18的合成:将化合物16b(3.6g,10.0mmol)、化合物8a(1.5g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物18)2.1g,收率为47.2%,esi(+) m/z=446.1。
[0061]
实施例191-(4-(3-(3-氯-5-氟吡啶-4-基)-1-甲基脲基)吡啶-2-基)-1,2,3,4-四氢喹啉-7-甲酰胺按照实施例16进行化合物16b的合成。
[0062]
化合物19的合成:将化合物16b(3.6g,10.0mmol)、化合物9a(1.8g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物19)2.6g,收率为55.3%,esi(+) m/z=471.1。
[0063]
实施例201-(2-(7-氰基-3,4-二氢喹啉-1(2h)-基)吡啶-4-基)-3-(3-氯-5-氟吡啶-4-基)-1-甲基脲
按照实施例16进行化合物16b的合成。
[0064]
化合物20的合成:将化合物16b(3.6g,10.0mmol)、化合物10a(1.6g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物20)2.8g,收率为61.9%,esi(+) m/z=453.1。
[0065]
实施例213-(2-氯-6-甲氧基苯基)-1-(2-(6-甲氧基吲哚啉-1-基)吡啶-4-基)-1-甲基脲化合物21b的合成:将化合物21a(18.3g,100.0mmol)、1b(18.6g,100.0mmol)、碳酸钾(20.7g,150.0mmol)溶于乙腈(300ml)中,室温反应10小时,tlc监测反应,反应完毕后过滤,滤液浓缩、柱层析分离得到类白色(化合物21b)28.0g,收率为75.9%。
[0066]
化合物21的合成:将化合物21b(3.7g,10.0mmol)、化合物1d(1.5g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物21)2.4g,收率为54.8%,esi(+) m/z=439.1。
[0067]
实施例223-(2-氯-6-甲氧基苯基)-1-(2-(6-氟吲哚啉-1-基)吡啶-4-基)-1-甲基脲按照实施例21进行化合物21b的合成。
[0068]
化合物22的合成:将化合物21b(3.7g,10.0mmol)、化合物2a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物22)2.7g,收率为63.4%,esi(+) m/z=427.1。
[0069]
实施例233-(2-氯-6-甲氧基苯基)-1-(2-(7-氟吲哚啉-1-基)吡啶-4-基)-1-甲基脲
按照实施例21进行化合物21b的合成。
[0070]
化合物23的合成:将化合物21b(3.7g,10.0mmol)、化合物3a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物23)3.0g,收率为70.4%,esi(+) m/z=427.1。
[0071]
实施例241-(4-(3-(2-氯-6-甲氧基苯基)-1-甲基脲基)吡啶-2-基)吲哚啉-6-甲酰胺按照实施例21进行化合物21b的合成。
[0072]
化合物24的合成:将化合物21b(3.7g,10.0mmol)、化合物4a(1.6g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物24)2.6g,收率为57.6%,esi(+) m/z=452.1。
[0073]
实施例253-(2-氯-6-甲氧基苯基)-1-(2-(6-氰基吲哚啉-1-基)吡啶-4-基)-1-甲基脲按照实施例21进行化合物21b的合成。
[0074]
化合物25的合成:将化合物21b(3.7g,10.0mmol)、化合物5a(1.4g,10.0mmol)、碳酸铯(6.5g,20.0mmol)、pd2(dba)3(458mg,0.5mmol)、xantphos(578mg,1.0mmol)溶于dmf(50ml),升温至100℃搅拌反应6小时,tlc监测反应,反应完毕后加水淬灭反应,乙酸乙酯(每次50ml)两次萃取,有机层浓缩,柱层析分离得到类白色固体(化合物25)2.3g,收率为53.1%,esi(+) m/z=434.1。
[0075]
实施例26fgfr4抑制活性测试化合物溶液:化合物在100%dmso中溶解稀释至100um,然后用dmso进行4倍的系列稀释
至8个不同的溶度,备用。
[0076]
反应缓冲液:激酶反应缓冲液(50mm hepes,ph7.0)、5mm氯化镁(mgcl2)、1mm二硫苏糖醇(dtt)和人重组fgfr4催化结构域蛋白。
[0077]
底物反应溶液:用反应缓冲液稀释成1000nm的生物素标记的酪氨酸激酶底物和90um三磷酸腺苷(atp)。
[0078]
检测液:0.125ng/ul eu 3+标记的笼状抗体和62.5nm链霉亲和素标记xl665。
[0079]
向384孔检测板中添加化合物溶液和fgfr4激酶溶液,混合均匀后室温孵育30分钟,随后加入底物反应液溶液,将反应混合物在室温孵育60分钟,随后加入与反应等体积检测液,混合均匀后室温放置。60分钟后,用乙二胺四乙酸溶液终止酶反应,envision检测,得到各个化合物的fgfr4活性(未加fgfr4蛋白组作为阴性对照,加fgfr4蛋白但是未加化合物组作为阳性对照),化合物的ic
50
通过软件计算而得到。
[0080]
激酶试验测试结果:a<100nm,100 nm≤b≤1000 nm,1000 nm ≤c
[0081]
实验结果表明:7个化合物(化合物4、化合物5、化合物8、化合物12、化合物17、化合物18、化合物23)对脾酪氨酸激酶抑制活性小于100nm。
[0082]
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips