
 商标分类
商标分类  商标转让
商标转让 
高产雅槛蓝醇型倍半萜的酿酒酵母工程菌及其构建方法与应用与流程
 2021-02-01 23:02:46|
2021-02-01 23:02:46| 330|
330| 起点商标网
起点商标网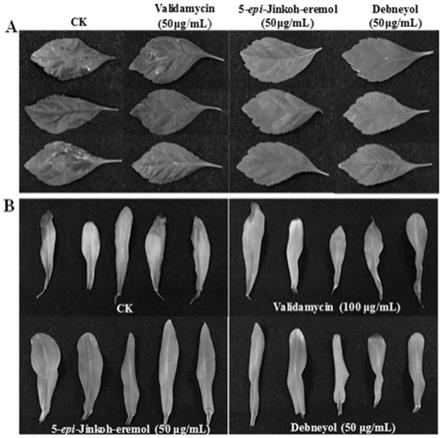
[0001]
本发明涉及代谢工程和植物抗病领域,特别涉及一种高产雅槛蓝醇型倍半萜的酿酒酵母工程菌及其构建方法与应用。
背景技术:
[0002]
植物生长的环境中存在大量的病原微生物,其中的真菌类病原菌往往给植物的生长和产量产生严重的威胁,特别是在农业和中草药种植领域,真菌性病害造成的损失不可估量。立枯丝核菌(rhizoctonia solani)为无孢科、丝核菌属半知菌真菌,是一种土传病原菌,可引起很多农作物的枯萎病以及中药材的立枯病,包括水稻(kouzai y,kimura m,watanabe m et al.salicylic acid-dependent immunity contributes to resistance against rhizoctonia solani,a necrotrophic fungal agent of sheath blight,in rice and brachypodium distachyon.new phytologist,2008,217:771-783)、玉米(gonz
á
lez-vera ad,bernardes-de-assis j,zala m et al.divergence between sympatric rice-and maize-infecting populations of rhizoctonia solani ag-1ia from latin america.phytopathology,2010,100:172-182)、半夏(范刚强,金义兰,桑维钧等.贵州省半夏常见病害种类调查与鉴定初报.山地农业生物学报,2009,28:312-316)、白车轴草(孙慧颖,杨丽娜,谢昀烨,万丹凤等.白车轴草夏枯病菌的生物学特性及药剂敏感性.吉林农业大学学报,2016,38:151-157)、红景天(白庆荣,谢昀烨,姜旭宇等.高山红景天立枯病菌菌丝融合群鉴定及药剂敏感性.东北林业大学学报,2014,1:107-111)等。尖孢镰刀菌(fusarium oxysporum)是一种世界性分布的病原真菌,寄主范围广泛,被列为植物十大真菌病害之一(dean r,van kan jal,pretorius za et al.the top 10fungal pathogens in molecular plant pathology.molecular plant pathology,2012,13:414-430),可引起农作物的枯萎病和中药材的根腐病,包括棉花(liu n,zhang x,sun y et al.molecular evidence for the involvement of a polygalacturonase-inhibiting protein,ghpgip1,in enhanced resistance to verticillium and fusarium wilts in cotton.scientific reports,2017,7:39840)、亚麻(dmitriev aa,krasnov gs,rozhmina ta et al.differential gene expression in response to fusarium oxysporum infection in resistant and susceptible genotypes of flax(linum usitatissimum l.).bmc plant biology,2017,17:253)、太子参(林茂兹,黄少华,陈巧巧等.太子参连作障碍及其根际土壤尖孢镰刀菌数量变化.云南农业大学学报,2012,27:716-721)、丹参(杨立,缪作清,杨光等.丹参枯萎病及其病原菌的研究.中国中药杂志,2013,38:4040-4043)等也都很容易被尖孢镰刀菌侵染,引起根腐病。由此可见,在农业和中草药种植行业,加强对病原真菌的防治是至关重要的。
[0003]
植物源农药是来源于植物体的农药,包括植物自身以及从中提取的活性成分和按活性结构合成的化合物及衍生物(张鹏,李西文,董林林等.植物源农药研发及中药材生产
中的应用现状.中国中药杂志,2016,41:3579-3586),是极具有潜力和市场的生物农药。我国药用植物资源丰富,品种繁多,很多中草药中含有抑制植物病原真菌的活性化合物,是开发植物源杀真菌农药的理想来源。迄今为止,已经有许多学者在药用植物抑制植物病原真菌方面进行了大量的研究,如银杏的种皮提取物能够对水稻纹枯病的致病真菌立枯丝核菌产生抗性(oh ts,koo hm,yoon hr et al.antifungal action of ginkgo biloba outer seedcoat on rice sheath blight.plant pathology journal,2016,31:61-66);灵芝的甲醇提取物能够抑制链格孢霉菌(aspergillus niger)、青霉菌(penicillium sp.)、黑曲霉菌、尖孢镰刀菌的生长(baig mn,shahid aa,ali m.in vitroassessment of extracts of the lingzhi or reishi medicinal mushroom,ganoderma lucidum(higher basidiomycetes)against different plant pathogenic fungi.international journal of medicine mushrooms,2015,17:407-11);续断的根中分离得的甾醇、皂苷等化合物能够对包括稻瘟病菌、立枯丝核菌、灰霉菌、炭疽病菌在内的真菌病原菌产生抗性,其中的皂苷类物质被认为可以作为抗真菌的先导化合物(choi nh,jang jy,choi gj et al.antifungal activity of sterols and dipsacus saponins isolated from dipsacus asper roots against phytopathogenic fungi.pesticide biochemistry and physiology,2017,141:103-108)。
[0004]
目前,针对农作物和中药材的真菌性病害的防止主要依靠化学农药,但化学农药环境污染严重,农药残留严重,制约着我国粮食的生产安全以及中药的国际化进展。因此,开发使用具有绿色环保、无残留的植物源农药成为必然的选择。
技术实现要素:
[0005]
本发明的首要目的在于克服现有技术的缺点与不足,提供一种高产雅槛蓝醇型倍半萜的酿酒酵母工程菌。
[0006]
本发明的另一目的在于提供所述高产雅槛蓝醇型倍半萜的酿酒酵母工程菌的构建方法。
[0007]
本发明的又一目的在于提供所述高产雅槛蓝醇型倍半萜的酿酒酵母工程菌的应用。
[0008]
本发明的目的通过下述技术方案实现:一种高产雅槛蓝醇型倍半萜的酿酒酵母工程菌,为菌株cr-1和cr-2中的至少一种;
[0009]
所述菌株cr-1以酿酒酵母为出发菌株,敲除lpp1、dpp1和gdh1基因,过表达erg8、erg10、thmg1、erg12、erg13、erg19、idi1、upc2-1、crtps18和smfpps基因;其中,
[0010]
thmg1和upc2-1基因整合到酿酒酵母染色体ty3位点;
[0011]
idi1和smfpps基因整合到酿酒酵母染色体ty4位点;
[0012]
erg8、erg10、erg12、erg13和erg19基因整合到酿酒酵母染色体his3位点;
[0013]
crtps18基因整合到酿酒酵母染色体ndt80位点;
[0014]
所述菌株cr-2以菌株cr-1为出发菌株,将crcyp71d349和atcpr1整合到酿酒酵母染色体gal80位点。
[0015]
所述的酿酒酵母优选为酿酒酵母by4741。
[0016]
所述敲除lpp1、dpp1和gdh1基因为运用crispr-cas9基因敲除系统进行基因敲除,
具体步骤如下:
[0017]
(a)将lpp1、dpp1和gdh1基因的crrna spacer核酸序列seq id no.24通过限制性内切酶bsaι连接到pcrct载体上,得到重组质粒pcrct-1;
[0018]
(b)将重组质粒pcrct-1转化酿酒酵母,经过培养筛选,得到敲除lpp1、dpp1和gdh1基因的菌株。
[0019]
步骤(b)中所述的酿酒酵母优选为酿酒酵母by4741。
[0020]
步骤(b)中所述的培养的温度优选为30℃。
[0021]
步骤(b)中所述的筛选为通过sd-ura缺陷培养基进行筛选。
[0022]
所述的erg8、erg10、thmg1、erg12、erg13、erg19和idi1基因可从酿酒酵母by4741基因组克隆得到,其核苷酸序列分别如seq id no.1~7所示。
[0023]
所述的upc2-1基因为upc2基因(seq id no.8)第888位的gly突变为asp得到的upc2-1基因,其核苷酸序列如seq id no.19所示。
[0024]
所述的crtps18基因为以长春花cdna为模板克隆得到的crtps18基因,其核苷酸序列如seq id no.20所示。
[0025]
所述的crcyp71d349基因为以长春花cdna为模板克隆得到的crcyp71d349基因,其核苷酸序列如seq id no.21所示。
[0026]
所述的atcpr1基因为以拟南芥cdna为模板克隆得到的atcpr1基因,其核苷酸序列如seq id no.22所示。
[0027]
所述的smfpps基因为以丹参cdna为模板克隆得到的smfpps基因,其核苷酸序列如seq id no.23所示。
[0028]
所述的高产雅槛蓝醇型倍半萜的酿酒酵母工程菌的构建方法,包括如下步骤:
[0029]
(1)基因敲除:
[0030]
将lpp1、dpp1和gdh1基因的crrna spacer核酸序列seq id no.24通过限制性内切酶bsaι连接到pcrct载体上,得到重组质粒pcrct-1;然后将重组质粒pcrct-1转化酿酒酵母by4741,经过培养筛选,得到敲除lpp1、dpp1和gdh1基因的菌株by4741-1;
[0031]
(2)利用重叠pcr构建以下10个模块:
[0032]
(a)将erg8、p
tdh2
和t
pyx212
重叠,构建基因表达模块p
tdh2-erg8-t
pyx212
,命名为模块i;
[0033]
(b)将erg10、p
pgk1
和t
adh1
重叠,构建基因表达模块p
pgk1-erg10-t
adh1
,命名为模块ii;
[0034]
(c)将erg12、p
tdh3
和t
tdh2
重叠,构建基因表达模块p
tdh3-erg12-t
tdh2
,命名为模块iii;
[0035]
(d)将erg13、p
tef1
和t
cyc1
重叠,构建基因表达模块p
tef1-erg13-t
cyc1
,命名为模块iv;
[0036]
(e)将erg19、p
tpi1
和t
fba1
重叠,构建基因表达模块p
tpi1-erg19-t
fba1
,命名为模块v;
[0037]
(f)将crtps18、p
tpi1
和t
fba1
重叠,构建基因表达模块p
tpi1-crtps18-t
fba1
,命名为模块vi;
[0038]
(g)将crcyp71d349、p
pgk1
和t
adh1
重叠,构建基因表达模块p
pgk1-crcyp71d349-t
adh1
,命名为模块vii;
[0039]
(h)将atcpr1、p
tef1
和t
cyc1
重叠,构建基因表达模块p
tef1-atcpr1-t
cyc1
,命名为模块
viii;
[0040]
(i)将thmg1、p
pgk1
、p
tef1
和upc2-1重叠,构建基因表达模块thmg1-p
pgk1-p
tef1-upc2-1,命名为模块ix;
[0041]
(j)将idi1、smfpps、p
pgk1
、p
tef1
重叠,构建基因表达模块idi1-p
pgk1-p
tef1-smfpps,命名为模块x;
[0042]
(3)构建菌株by4741-5
[0043]
(i)将步骤(i)中得到的模块ix插入到载体pcfb2875的sfaι酶切位点,得到整合表达载体pcfb2875-1;然后用限制性核酸内切酶notι酶切整合表达载体pcfb2875-1,得到dna整合片段a1;再将dna整合片段a1整合到步骤(1)中得到的菌株by4741-1的染色体ty3位点,得到菌株by4741-2;
[0044]
(ii)将步骤(j)中得到的模块x插入到载体pcfb2798的sfaι酶切位点,得到整合表达载体pcfb2798-1;然后用限制性核酸内切酶notι酶切整合表达载体pcfb2798-1,得到dna整合片段a2;再将dna整合片段a2整合到步骤(i)中得到的菌株by4741-2的染色体ty4位点,得到菌株by4741-3;
[0045]
(iii)将psh65载体转化菌株by4741-3,然后用含有zeocin(博来霉素)的ypd培养基进行筛选(筛选kileu2和kiura3都被敲除的菌株),得到菌株by4741-4;
[0046]
(iv)将模块i、ii、iii、iv、v和筛选标记met15共同转化菌株by4741-4(即利用筛选标记met15将模块vi和vii整合到菌株by4741-4基因组的his3位点),得到菌株by4741-5;
[0047]
(4)构建菌株cr-1
[0048]
将模块vi和筛选标记his3共同转化菌株by4741-5,然后通过酵母缺陷型培养基sd-his筛选培养(即利用筛选标记his3将模块vi整合到菌株by4741-5基因组的ndt80位点),得到菌株cr-1,即为所述的高产雅槛蓝醇型倍半萜的酿酒酵母工程菌;
[0049]
(5)构建菌株cr-2
[0050]
将模块vii、viii和筛选标记kiura3共同转化菌株cr-1,然后通过酵母缺陷型培养基sd-ura筛选培养(即利用筛选标记kiura3将模块vii和viii,整合到菌株cr-1基因组的gal80位点),得到菌株cr-2,即为所述的高产雅槛蓝醇型倍半萜的酿酒酵母工程菌。
[0051]
步骤(2)中所述的erg8、erg10、thmg1、erg12、erg13、erg19和idi1基因可从酿酒酵母by4741基因组克隆得到,其核苷酸序列分别如seq id no.1~7所示。
[0052]
步骤(2)中所述的p
pgk1
、p
tef1
、p
tdh3
、p
tdh2
和p
tpi1
为启动子序列,可从酿酒酵母by4741基因组克隆得到,其核苷酸序列分别如seq id no.9~13所示。
[0053]
步骤(2)中所述的t
adh1
、t
cyc1
、t
tdh2
、t
pyx212
和t
fba1
为终止子序列,可从酿酒酵母by4741基因组克隆得到,其核苷酸序列分别如seq id no.14~18所示。
[0054]
步骤(2)中所述的upc2-1基因为upc2基因第888位的gly突变为asp得到的upc2-1基因,其核苷酸序列如seq id no.19所示。
[0055]
步骤(2)中所述的crtps18基因为以长春花cdna为模板克隆得到的crtps18基因,其核苷酸序列如seq id no.20所示。
[0056]
步骤(2)中所述的crcyp71d349基因为以长春花cdna为模板克隆得到的crcyp71d349基因,其核苷酸序列如seq id no.21所示。
[0057]
步骤(2)中所述的atcpr1基因为以拟南芥cdna为模板克隆得到的atcpr1基因,其
jinkoh-eremol和debneyol在玉米上对玉米黑粉病菌的抗真菌效果图。
具体实施方式
[0090]
下面结合实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。下列实施例中未注明具体实验条件的试验方法,通常按照常规实验条件或按照制造厂所建议的实验条件。除非特别说明,本发明所用试剂和原材料均可通过市售获得。
[0091]
本发明实施例中所涉及的培养基的配方如下:
[0092]
(1)ypd培养基:蛋白胨20g/l,酵母提取物10g/l,葡萄糖20g/l(固体ypd培养基在配制时添加20g/l的琼脂粉)。
[0093]
(2)ypg培养基:蛋白胨20g/l,酵母提取物10g/l,葡萄糖2g/l,半乳糖18g/l。
[0094]
(3)sd-ura培养基:ynb培养基6.7g/l,ura(尿嘧啶)缺陷氨基酸(100x)10ml/l,葡萄糖20g/l(固体培养基配制时添加20g/l的琼脂粉)。
[0095]
(4)sd-met培养基:ynb培养基6.7g/l,met(甲硫氨酸)缺陷氨基酸(100x)10ml/l,葡萄糖20g/l(固体培养基配制时添加20g/l的琼脂粉)。
[0096]
(5)sd-leu培养基:ynb培养基6.7g/l,leu(亮氨酸)缺陷氨基酸(100x)10ml/l,葡萄糖20g/l(固体培养基配制时添加20g/l的琼脂粉)。
[0097]
(6)sd-his培养基:ynb培养基6.7g/l,his(组氨酸)缺陷氨基酸(100x)10ml/l,葡萄糖20g/l(固体培养基配制时添加20g/l的琼脂粉)。
[0098]
缺陷氨基酸母液(100x):腺嘌呤硫酸盐0.25g,精氨酸0.12g,天冬氨酸0.6g,谷氨酸0.6g,组氨酸0.12g,亮氨酸0.36g,赖氨酸0.18g,甲硫氨酸0.12g,苯丙氨酸0.3g,丝氨酸2.25g,苏氨酸1.2g,色氨酸0.24g,酪氨酸0.18g,缬氨酸0.9g,尿嘧啶0.12g,用ddh2o定容到57ml,根据需要可不加任意一种氨基酸配置成缺陷氨基酸母液(100x)。上述原料均购买自sigma-aldrich。
[0099]
(7)发酵培养基:25g/l葡萄糖,(nh4)2so
4 15g/l,kh2po
4 8g/l,mgso
4 3g/l,znso4·
7h2o 0.72g/l,维生素溶液12ml/l,以及微量金属盐溶液10ml/l。
[0100]
(8)补料培养基:葡萄糖585g/l,kh2po
4 9g/l,mgso
4 2.5g/l,k2so
4 3.5g/l,na2so
4 0.28g/l,维生素溶液12ml/l,以及微量金属盐溶液10ml/l;其中,
[0101]
维生素溶液:维生素h 0.05g/l,泛酸钙1g/l,烟酸1g/l,肌醇25g/l,盐酸硫胺素1g/l,盐酸吡哆醇1g/l和对氨基苯甲酸0.2g/l。
[0102]
微量金属盐溶液:edta(乙二胺四乙酸)15g/l,znso4·
7h2o 10.2g/l,mncl2·
4h2o 0.5g/l,cuso
4 0.5g/l,cocl2·
6h2o 0.86g/l,na2moo4·
2h2o 0.56g/l,cacl2·
2h2o 3.84g/l和feso4·
7h2o 5.12g/l。
[0103]
(9)pda培养基:马铃薯葡萄糖粉末6g/l(固体培养基配制时添加20g/l的琼脂粉)。
[0104]
(10)玉米黑粉菌培养基:马铃薯葡萄糖粉末6g/l,kh2po
4 3.0g/l,mgso4·
7h2o 1.5g/l(固体培养基配制时添加20g/l的琼脂粉)。
[0105]
实施例1酵母内源性基因、启动子、终止子和枯草芽孢杆菌gdh基因的克隆
[0106]
1、酵母基因组的提取
[0107]
(1)挑取酿酒酵母by4741(购于atcc)的单克隆于5ml ypd培养基中,30℃培养20~
24h,而后4000rpm离心5min,集菌,并置于研钵中。
[0108]
(2)液氮速冻,研磨,待液氮挥干后,加入1ml dnaiso reagent(宝日医生物技术有限公司),混匀。
[0109]
(3)将裂解液转移至离心管中,于4℃或室温12,000rpm离心10min。
[0110]
(4)将上清液转移到新的离心管内,加入1/2体积的无水乙醇,混匀,室温4000rpm离心,去上清。
[0111]
(5)用75%(v/v)的乙醇清洗沉淀2次,并挥干残留乙醇,加入50μl ddh2o溶解,作为pcr克隆模板。
[0112]
2、酵母内源性基因和表达元件的克隆
[0113]
(1)以上述获得的酵母基因组为模板,分别克隆以下7个基因、5个启动子和5个终止子(扩增引物见表1):
[0114]
erg8(引物erg8-f、erg8-r)、erg10(引物erg10-f、erg10-r)、thmg1(引物thmg1-f、thmg1-r)、erg12(引物erg12-f、erg12-r)、erg13(引物erg13-f、erg13-r)、erg19(引物erg19-f、erg19-r),idi1(引物idi1-f、idi1-r)和upc2(引物upc2-f、upc2-r)基因;其中,erg8、erg10、thmg1、erg12、erg13、erg19、idi1和upc2的基因序列分别如seq id no.1~8所示。
[0115]
p
pgk1
(引物ppgk1-f、ppgk1-r)、p
tef1
(引物ptef1-f、ptef1-r)、p
tdh3
(引物ptdh3-f、ptdh3-r)、p
tdh2
(引物ptdh2-f、ptdh2-r)和p
tpi1
(引物ptpi1-f、ptpi1-r)启动子,其核苷酸序列分别如seq id no.9~13所示。
[0116]
t
adh1
(引物tadh1-f、tadh1-r)、t
cyc1
(引物tcyc1-f、tcyc1-r)、t
tdh2
(引物ttdh2-f、ttdh2-r)、t
pyx212
(引物tpyx212-f、tpyx212-r)和t
fba1
(引物tfba1-f、tfba1-r)终止子,其核苷酸序列分别如seq id no.14~18所示。
[0117]
pcr反应体系:phanta max高保真酶(phanta max super-fidelity dna polymerase)0.5μl,dntp(10mm)0.5μl,2x phanta max buffer 10μl,上下游特异性引物各0.5μl,ddh2o 7μl,模板1μl,总反应体系20μl。
[0118]
pcr扩增反应条件为:95℃1min;95℃30s,50-60℃30s,72℃1-2min,30循环;72℃7min。
[0119]
dna片段纯化:pcr反应结束后,琼脂糖凝胶电泳检测条带,并使用普通琼脂糖凝胶dna回收试剂盒(天根生化科技有限公司)进行胶回收,具体操作过程见说明书。
[0120]
(2)dna片段连接peasy-blunt载体
[0121]
dna片段连接平末端peasy-blunt(全式金生物技术有限公司),转化dh5α菌株,具体操作见产品说明书。
[0122]
(3)37℃培养14~16h后,挑选单菌落做菌落pcr。
[0123]
pcr反应体系:easy taq聚合酶0.2μl,dntps(2.5mm)0.8μl,10x easy taq buffer 1μl,通用引物m13f(表1)(10μm)0.3μl,m13r(表1)(10μm)0.3μl,dmso 1μl,ddh2o 6.4μl。
[0124]
pcr扩增条件为:95℃5min;95℃30s,55℃30s,72℃1-3min,30个循环;72℃7min。
[0125]
反应结束后,琼脂糖凝胶电泳检测阳性转化子,送样测序。
[0126]
3、upc2的定点突变
[0127]
upc2是正向调控酵母mva途径的重要转录因子,可以促进萜类化合物的底物ipp和
dmapp的合成,将该转录因子的第888位的甘氨酸(glycine)突变为天门冬氨酸(aspartate),能进一步提高其调控效率。利用pcr的方法,以peasy-blunt-upc2质粒(peasy-blunt-upc2质粒由upc2基因构建到peasy-blunt载体上制备得到,具体参考实施例1)为模板,进行定点突变(引物序列见表1),具体步骤如下:
[0128]
(1)pcr反应系:phanta max super-fidelity dna polymerase 0.5μl,dntp 0.5μl,2x phanta max buffer 10μl,正向、反向引物各1μl(引物见表1:上游引物upc2-1-f和下游引物upc2-1-r),ddh2o 6μl,模板1μl,总反应体系20μl。
[0129]
(2)pcr反应条件:95℃3min;95℃30s,62℃30s,72℃2min,18个循环;72℃7min。
[0130]
(3)向pcr产物中加入0.5μl限制性内切酶dpnι,37℃消化1h。
[0131]
(4)将消化体系全部转化大肠杆菌dh5α,37℃倒置培养14~18h。
[0132]
(5)菌落pcr挑选阳性转化子进行测序。
[0133]
upc2定点突变得到的upc2-1的核苷酸序列如seq id no.19所示。
[0134]
实施例2、植物来源的基因的克隆
[0135]
1、植物组织rna的提取
[0136]
利用快速通用rna提取试剂盒(北京华越洋生物科技有限公司)提取长春花、丹参、拟南芥(长春花、丹参、拟南芥均可通过常规市售购买得到)叶片中的总rna,具体操作过程见说明书。
[0137]
2、mrna反转录成cdna
[0138]
采用hiscript ii reverse transcriptase试剂盒(南京诺维赞生物科技有限公司)将rna反转录成cdna,具体操作见试剂盒说明书。
[0139]
3、目的基因的克隆
[0140]
以长春花cdna为模板克隆crtps18和crcyp71d349基因,以拟南芥cdna为模板克隆atcpr1基因,以丹参cdna为模板克隆smfpps基因,引物见表1,具体实施方法可参考实施例1。克隆得到的crtps18、crcyp71d349、atcpr1和smfpps的基因序列如seq id no.20~23所示。
[0141]
表1.本发明涉及的克隆基因和表达元件所用引物序列
[0142]
[0143][0144]
实施例3、crispr/cas9技术敲除酿酒酵母中的目的基因
[0145]
1、目的基因spacer核酸序列的设计和载体的构建
[0146]
参考文献(bao z,xiao h,liang j et al.homology-integrated crispr
–
cas(hi-crispr)system for one-step multigene disruption in saccharomyces cerevisiae.acs synthetic biology,2015,5:585-594))中的crispr/cas9靶点设计原则,设计消耗法尼基焦磷酸(fpp)的lpp1和以及消耗辅酶nadph的dpp1基因,gdh1三个基因的spacer序列,并委托苏州泓迅生物科技有限公司合成。lpp1、dpp1、gdh1基因的crrna spacer核酸序列如seq id no.24所示,crrna spacer序列通过限制性内切酶bsaι酶切连接到pcrct载体(武汉淼灵生物科技有限公司)上。
[0147]
2、pcrct载体转化酿酒酵母敲除目的基因
[0148]
(1)利用zymo research frozen-ez yeast transformation ii kit
tm
酵母转化试剂盒(上海懋康生物科技有限公司)进行酵母转化,将pcrct载体转化酿酒酵母by4741,具体转化方法参照产品说明书,30℃培养4~5d(天)。
[0149]
(2)利用营养缺陷型培养基sd-ura和菌落pcr筛选阳性转化子。
[0150]
(3)将阳性转化子接种于4ml sd-ura培养基中,30℃、220rpm条件下培养2d。
[0151]
(4)取100μl菌液接于新鲜sd-ura培养基中培养2d。
[0152]
(5)将菌液稀释后涂于sd-ura缺陷平板上,挑选单克隆摇菌2d,离心后集菌,利用
dnaiso reagent基因组提取试剂盒提取基因组。
[0153]
(6)pcr克隆目的基因,产物直接测序,检测目的基因的敲除情况。
[0154]
(7)将已经敲除目的基因的菌株用ypd培养基传代培养十天,以去除pcrct载体,得到菌株by4741-1。
[0155]
实施例4、各表达模块的构建
[0156]
1、利用重叠pcr构建基因表达模块i~x
[0157]
(1)各模块的构建都通过重叠pcr技术,模块中各连接的片段通过pcr克隆,加上40~50bp的重叠区域,重叠区域碱基退火温度在60~70℃之间。
[0158]
(2)第一轮反应体系:phanta max super-fidelity dna polymerase 0.5μl,dntp(10mm)0.5μl,2x phanta max buffer 10μl,dna片段1~5μl,上下游引物各0.5μl(见表2),加ddh2o至20μl;其中引物序列见表2。
[0159]
第一轮pcr反应条件:95℃3min;95℃30s,60~70℃30s,72℃1~4min,15个循环;72℃7min。
[0160]
(3)取第一轮pcr反应液1μl作为第二轮pcr模板,第二轮pcr体系:phanta max super-fidelity dna polymerase 0.5μl,dntp(10mm)0.5μl,2x phanta max buffer 10μl,上下游引物各0.5μl(引物为连接成完整dna片段上下游引物,见表2),ddh2o 7μl,模板1μl,总反应体系20μl。
[0161]
第二轮pcr反应条件:95℃3min;95℃30s,50~60℃30s,72℃1~4min,30个循环;72℃5min。
[0162]
(4)胶回收并连接peasy-blunt载体(全式金生物技术有限公司)测序。
[0163]
按上述步骤共构建以下10个模块:
[0164]
(a)将erg8、p
tdh2
和t
pyx212
重叠,构建基因表达模块p
tdh2-erg8-t
pyx212
,命名为模块i;其中,第一轮pcr克隆erg8引物为i-erg8-f、i-erg8-r,克隆p
tdh2
引物为p
tdh2-f、i-p
tdh2-r,克隆t
pyx212
引物为i-t
pyx212-f、t
pyx212-r;第二轮pcr引物为p
tdh2-f、t
pyx212-r;
[0165]
(b)将erg10、p
pgk1
和t
adh1
重叠,构建基因模块p
pgk1-erg10-t
adh1
,命名为模块ii;其中,第一轮pcr克隆erg10引物为ii-erg10-f、ii-erg10-r,克隆p
pgk1
引物为p
pgk1-f、ii-p
pgk1-r,克隆t
adh1
引物为ii-t
adh1-f、t
adh1-r;第二轮pcr引物为p
pgk1-f、t
adh1-r;
[0166]
(c)将erg12、p
tdh3
和t
tdh2
重叠,构建基因表达模块p
tdh3-erg12-t
tdh2
,命名为模块iii;其中,第一轮pcr克隆erg12引物为iii-erg12-f、iii-erg12-r,克隆p
tdh3
引物为p
tdh3-f、iii-p
tdh3-r,克隆t
tdh2
引物为iii-t
tdh2-f、t
tdh2-r;第二轮pcr引物为p
tdh3-f、t
tdh2-r;
[0167]
(d)将erg13、p
tef1
和t
cyc1
重叠,构建基因表达模块p
tef1-erg13-t
cyc1
,命名为模块iv;其中,第一轮pcr克隆erg13引物为iv-erg13-f、iv-erg13-r,克隆p
tef1
引物为p
tef1-f、iv-p
tef1-r,克隆t
cyc1
引物为iv-t
cyc1-f、t
cyc1-r;第二轮pcr引物为p
tef1-f、t
cyc1-r;
[0168]
(e)将erg19、p
tpi1
和t
fba1
重叠,构建基因表达模块p
tpi1-erg19-t
fba1
,命名为模块v;其中,第一轮pcr克隆erg19引物为v-erg19-f、v-erg19-r,克隆p
tpi1
引物为p
tpi1-f、v-p
tpi1-r,克隆t
fba1
物为v-t
fba1-f、t
fba1-r;第二轮pcr引物为p
tpi1-f、t
fba1-r;
[0169]
(f)将crtps18、p
tpi1
和t
fba1
重叠,构建基因表达模块p
tpi1-crtps18-t
fba1
,命名为模块vi;其中,第一轮pcr克隆crtps18引物为vi-crtps18-f、vi-crtps18-r,克隆p
tpi1
引物为p
tpi1-f、vi-p
tpi1-r,克隆t
fba1
物为vi-t
fba1-f、t
fba1-r;第二轮pcr引物为p
tpi1-f、t
fba1-r;
[0170]
(g)将crcyp71d349、p
pgk1
和t
adh1
重叠,构建基因表达模块p
pgk1-crcyp71d349-t
adh1
,命名为模块vii;其中,第一轮pcr克隆crcyp71d349引物为vii-crcyp71d349-f、vii-crcyp71d349-r,克隆p
pgk1
引物为p
pgk1-f、vii-ppgk1-r,克隆t
adh1
物为ii-t
adh1-f、tadh
1-r;第二轮pcr引物为p
pgk1-f、t
adh1-r;
[0171]
(h)将atcpr1、p
tef1
和t
cyc1
重叠,构建基因表达模块p
tef1-atcpr1-t
cyc1
,命名为模块viii;其中,第一轮pcr克隆atcpr1引物为viii-atcpr1-f、viii-atcpr1-r,克隆p
tef1
引物为p
tef1-f、viii-p
tef1-r,克隆t
cyc1
引物为viii-t
cyc1-f、t
cyc1-r;第二轮pcr引物为p
tef1-f、t
cyc1-r;
[0172]
(i)将thmg1、p
pgk1
、p
tef1
和upc2-1重叠,构建基因表达模块thmg1-p
pgk1-p
tef1-upc2-1,命名为模块ix;其中,第一轮pcr克隆thmg1引物为ix-thmg1-f、ix-thmg1-r,克隆p
pgk1
引物为ix-p
pgk1-f、ix-p
pgk1-r,克隆p
tef1
引物为ix-p
tef1-f、ix-p
tef1-r,克隆upc2-1引物为ix-upc2-1-f、ix-upc2-1-r,第二轮pcr引物为ix-thmg1-f、ix-upc2-1-r;
[0173]
(j)将idi1、smfpps、p
tdh2
、p
tdh3
重叠,构建基因表达模块idi1-p
pgk1-p
tef1-smfpps,命名为模块x;其中,第一轮pcr克隆idi1引物为x-idi1-f、x-idi1-r,克隆p
tdh2
引物为x-p
tdh2-f、x-p
tdh2-r,克隆p
tdh3
引物为x-p
tdh3-f、x-p
tef1-r,克隆smfpps引物为x-smfpps-f、x-smfpps-r,第二轮pcr引物为x-idi1-f、ix-smfpps-r。
[0174]
表2构建基因表达模块所用引物序列
[0175]
[0176][0177]
2、整合载体pcfb2875-1和pcfb2798-1的构建
[0178]
(1)利用限制性内切酶sfaai线性化载体pcfb2875(addgene),酶切体系:pcfb2875载体1μg,sfaai 0.5μl,10x fast digest buffer 2μl,加水补齐到20μl体系。37℃酶切1h,胶回收线性化载体。
[0179]
(2)将模块ix从peasy-blunt-thmg1-p
pgk1-p
tef1-upc2-1(即上述模块ix连接peasy-blunt载体获得)上用sfaai切下来,酶切体系同载体pcfb2875线性化酶切体系。
[0180]
(3)将模块ix通过t4连接酶连接到载体pcfb875sfaι酶切位点,连接体系:线性化的pcfb2875 20~100ng,模块ix 50~500ng,t4dna ligase 0.5μl,10x t4dna ligase buffer 1μl,加水补齐到10μl体系。
[0181]
(4)16℃连接1h,将连接产物转化dh5α感受态细胞,37℃培养14~16h,通过菌落pcr筛选和提取质粒测序,获得连接好的载体pcfb2875-1。
[0182]
(5)含有模块x的整合表达载体pcfb2798-1的构建方法如pcfb2875-1。
[0183]
实施例5、构建工程酵母菌
[0184]
1、by4741-3菌株的构建
[0185]
(1)用限制性内切酶notι将包含模块ix的t
cyc1-ix-t
adh1
表达单元和筛选标记kiura3(kiura3的核苷酸序列如seq id no.25所示,由苏州泓迅生物科技有限公司合成)的dna整合片段从载体pcfb2875-1(实施例4步骤2构建)切下来。酶切体系:载体pcfb2875-1 3~5μg,notι限制性内切酶1~2μl,10x fast digest buffer 5μl,补水至50μl,37℃酶切1~2h。跑电泳检测酶切效果,并纯化。
[0186]
(2)取5μl上述纯化的dna片段,转化酿酒酵母by4741-1(方法同实施例3步骤2),将模块整合到酵母by4741-1染色体的ty3位点,用尿嘧啶缺陷型培养基(sd-ura培养基)平板进行筛选,得到工程菌株by4741-2。
[0187]
(3)用限制性内切酶notι将包含模块x的t
cyc1-x-t
adh1
表达单元和筛选标记kileu2(kileu2的核苷酸序列如seq id no.26所示,由苏州泓迅生物科技有限公司合成)的dna整合片段从载体pcfb2798-1(实施例4步骤2构建)切下来,并整合到酵母菌株by4741-2染色体ty4位点,整合方法同上所述,利用leu营养缺陷型培养基(sd-leu培养基)平板筛选,得到工程菌株by4741-3。
[0188]
2、敲除筛选标记
[0189]
(1)将psh65载体(武汉淼灵生物科技有限公司)转化by4741-3菌株(zymo research frozen-ez yeast transformation ii kit
tm
酵母转化试剂盒;上海懋康生物科技有限公司),用含有zeocin(100μg/ml)的ypd培养基筛选。
[0190]
(2)挑选单菌落用含有zeocin(100μg/ml)的ypd培养基培养四天,通过划板稀释挑选单菌落,提取基因组,通过pcr克隆出整合的dna片段,通过测序筛选得到kileu2和kiura3都被敲除的菌株(通过cre/loxp体系酶敲除kileu2和kiura3筛选标记),再用ypd培养基传代培养十天,以丢掉psh65质粒,得到工程菌株by4741-4。
[0191]
3、酵母菌株by4741-5的构建
[0192]
(1)利用酵母体内同源重组的方法,将模块i、ii、iii、iv和v整合到工程菌株by4741-4染色体的his3位点,通过pcr给相邻模块和筛选标记met15(核苷酸序列如seq id no.27所示,由苏州泓迅生物科技有限公司合成)加上50bp的重叠区域,同时在模块1的5'端和met15的3'端加上50bp同源臂用于整合到his3位点,整合顺序为模块i-ii-iii-iv-v-met15(即将这5个模块和筛选标记met15一起转化到酿酒酵母内会发生体内同源重组连接成完整的一条dna片段,并且由于两端的模块i和met15带有50bp特异性的同源臂,使得这个dna片段可以整合到酵母染色体的his3位点)。其中,克隆模块i的引物为his3-1-f、his3-1-r,克隆模块ii的引物为ppgk1-f、his3-2-r,克隆模块iii引物为ptdh3-f、his3-3-r,克隆模块iv的引物为ptef1-f、his3-4-r,克隆模块v的引物为ptpi1-f、his3-5r,克隆met15的引物为his3-met15(kileu2,his3,kiura3)-f、his3-met15-r。
[0193]
(2)将上述6个片段转化工程菌株by4741-4,用sd-met缺陷型培养基筛选和菌落pcr鉴定,得到阳性菌株by4741-5。
[0194]
4、5-epi-jinkoh-eremol高产酿酒酵母工程菌株cr-1的构建
[0195]
将模块vi整合到by4741-5菌株染色体ndt80位点上,所用筛选标记his3(his3的核苷酸序列如seq id no.28所示,由苏州泓迅生物科技有限公司合成),利用his缺陷型培养基(sd-his培养基)进行筛选,得到菌株cr-1。克隆模块vi的引物为ndt80-8-f、ndt80(gal80)-8-r,克隆模块ix的引物为p
pgk1-f、ndt80(gal80)-9-r,克隆模块x的引物为p
tef1-f、
ndt80(gal80)-10-r,克隆筛选标记his3的引物为his3-met15(kileu2,his3,kiura3)-f、ndt80-his3-r。克隆方法参考实施例5步骤3,构建菌株cr-1。
[0196]
5、debneyol高产酿酒酵母工程菌株cr-2的构建
[0197]
利用kiura3(kiura3的核苷酸序列如seq id no.27所示,由苏州泓迅生物科技有限公司合成)筛选标记第二次将模块vii和viii整合到cr-1菌株染色体gal80位点上,利用ura缺陷型培养基(sd-ura培养基)进行筛选。克隆模块vii的引物为ppgk1-f、ndt80(gal80)-9-r,克隆模块viii的引物为ptef1-f、ndt80(gal80)-10-r,克隆kiura3的引物为his3-met15(kileu2,his3,kiura3)-f、gal80-kiura3-r。方法参考实施例5步骤3(酵母菌株by4741-7的构建),构建菌株cr-2。
[0198]
表3.模块整合酵母染色体引物
[0199]
[0200][0201]
实施例6、菌株cr-1、cr-2发酵生成雅槛蓝醇型倍半萜
[0202]
1、菌株cr-1发酵生成5-epi-jinkoh-eremol
[0203]
(1)将菌株cr-1的单菌落接种到15ml的ypd培养基中,30℃220rpm培养od值至2~3。
[0204]
(2)将菌液接种到3瓶100ml的发酵培养基中,每瓶接种5ml,30℃220rpm培养od值至8~10。
[0205]
(3)将上述3瓶100ml菌液合并,都接种到3l的发酵培养基中进行发酵。发酵条件为:温度30℃,转速300~1000rpm,ph5.0(用氨水调节),溶氧值30%,通气量3~20l/min;当溶氧值达到60%时,启动补料系统进行补料,使培养基中葡萄糖的含量维持在5g/l;发酵时间为168h。
[0206]
(4)菌株cr-1发酵生成debneyol的方法与菌株cr-1发酵生成5-epi-jinkoh-eremol相同。
[0207]
2、菌株cr-2发酵生成5-epi-jinkoh-eremol和debneyol:方法同上。
[0208]
3、发酵产物的萃取、检测及纯化
[0209]
(1)取上述发酵液加入等体积的正己烷或乙酸乙酯超声萃取20min,静置24h,取200μl有机层于液相小瓶中,用安捷伦气质联用仪7890b-5977b进行gc-ms检测。检测方法:hp-5ms毛细管柱;进样量1μl,不分流;进样口温度250℃,起始温度50℃保持3min,而后以20℃/min的速率升到70℃保持1min,接着15℃/min的速率升到300℃保持3min;electron ionization离子源,能量强度70ev;ms溶剂延迟设为12min,开启电压倍增模式,增益因子设为1。离子检测设为离子选择模式(sim)并检测m/z 189,136,119,105等离子。
[0210]
(2)将剩余上述萃取的有机层旋转蒸发,浓缩至干燥,复溶于少量正己烷中,然后按1:40的比例过200~300目硅胶柱。柱纯化条件为:6倍柱体积的正己烷,6倍柱体积的正己烷/丙酮(40:1),3倍柱体积的正己烷/丙酮(30:1),3倍柱体积的正己烷/丙酮(20:1),3倍柱体积的正己烷/丙酮(10:1)。洗脱期间,利用薄层层析(tlc)检测目标产物,将含有目标产物的馏分经气相色谱分析后合并浓缩,并用高效液相色谱(hplc)进一步分离纯化。最后,将含有目标产物的馏分合并,用氮气吹干备用。
[0211]
(3)结果:酿酒酵母中重建雅槛蓝醇型倍半萜的生物合成途径如图1所示;酵母菌株发酵产物的gc-ms总离子流图和质谱图如图2所示;cr-1和cr-2菌株高密度发酵生产5-epi-jinkoh-eremol和debneyol的产量如图3所示:5-epi-jinkoh-eremol由crcyp71d349基因进一步氧化产生debneyol,cr1菌株没有crcyp71d349基因,故只产生5-epi-jinkoh-eremol;cr2菌株引入了crcyp71d349基因,故5-epi-jinkoh-eremol基本都全被转化为debneyol。
[0212]
实施例7、雅槛蓝醇型倍半萜抗植物病原真菌的应用
[0213]
1、植物病原真菌的活化和雅槛蓝醇型倍半萜母液的配置
[0214]
(1)利用马铃薯葡萄糖固体培养基(pda)和玉米黑粉病菌培养基活化植物病原真菌立枯丝核菌rhizoctonia solani和玉米黑粉病菌ustilago maydis(两种菌株均购自中国普通微生物菌种保藏中心,cgmcc)。
[0215]
(2)将菌株cr1发酵得到的5-epi-jinkoh-eremol和菌株cr2发酵得到的debneyol分别用dmso(二甲基亚砜)配成100mg/ml的母液,,同时将抗植物真菌的阳性药validamycin用dmso配成50mg/ml的母液。
[0216]
2、雅槛蓝醇型倍半萜在刺五加上对立枯丝核菌的抗性应用
[0217]
(1)将5-epi-jinkoh-eremol、debneyol和validamycin母液用无菌蒸馏水稀释成50μg/ml的工作液,并均匀喷洒在刺五加活体植株叶片表面。
[0218]
(2)用打孔器从活化立枯丝核菌的pda培养基上取直径为5mm的菌饼,将菌饼放到喷洒化合物的刺五加叶片上。将接种后的刺五加置于光照培养箱中,培养箱条件为:16h光照/8h黑暗,25℃,湿度70-80%。
[0219]
(3)48h后,统计并比较接种叶片上的病斑面积,以评估抗病效果。结果如图4a所示,对照组和50μg/ml的阳性药validamycin处理的刺五加叶片被立枯丝核菌侵染,并产生坏死斑;同浓度的5-epi-jinkoh-eremol和debneyol处理的刺五加叶片没有被侵染,无坏死斑的产生。这表明两种雅槛蓝醇型倍半萜对腐生营养型病原真菌立枯丝核菌能够产生抗病性。
[0220]
3、雅槛蓝醇型倍半萜在玉米上对玉米黑粉病菌的抗性应用
[0221]
(1)将5-epi-jinkoh-eremol、debneyol和validamycin母液用无菌蒸馏水稀释成50μg/ml的工作液,并均匀喷洒在玉米(三叶一心苗期)叶片表面。
[0222]
(2)从活化玉米黑粉病菌的培养基上挑取部分菌落,接种于液体玉米黑粉病专用培养基中,30℃,200rpm培养至od值为1。4500rpm离心集菌,用等体积的无菌蒸馏水重悬。
[0223]
(3)用1ml无菌注射器(带针头)将重悬的玉米黑粉病菌注射玉米第一片叶下的茎部,接种后,置于光照培养箱中,培养箱条件为:16h光照/8h黑暗,25℃,湿度70-80%。
[0224]
(4)72h后,观察并比较玉米第一片叶上的病斑面积,以评估抗病效果。结果如图4b所示,对照组玉米叶片褪绿面积基本占据了整个叶片,而50μg/ml的5-epi-jinkoh-eremol和debneyol与100μg/ml的阳性药validamycin处理的玉米叶片褪绿面积明显很小,表明两种雅槛蓝醇型倍半萜对活体营养型病原真菌玉米黑粉病菌具有显著的抗病活性。
[0225]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips