
 商标分类
商标分类  商标转让
商标转让 
一种合成2-氟环丁基甲胺及其中间体的方法与流程
 2021-02-01 23:02:59|
2021-02-01 23:02:59| 177|
177| 起点商标网
起点商标网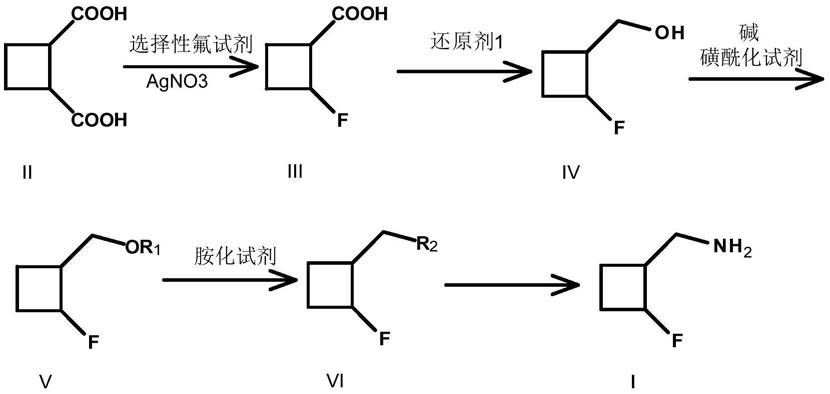
[0001]
本发明涉及药物中间体合成领域,具体地说涉及一种合成2-氟环丁基甲胺及其中间体的方法。
背景技术:
[0002]
2-氟环丁基甲胺是一个重要的医药中间体,在开发谷氨酸受体亚型5(mglur5)受体变构调节剂和一些抗肿瘤药物中都有应用。us2013345204a1报道了一部分包含2-氟环丁基甲胺结构片段的内酰胺类似物,作为mglur5受体的变构调节剂,这些类似物可以用于治疗或预防与谷氨酸功能失调相关的障碍和其中涉及受体的mglur5亚型的疾病,目前2-氟环丁基甲胺的合成方法没有文献报道。因此,需要开发一个原料易得,操作方便,反应易于控制,适合工业化生产的合成方法。
技术实现要素:
[0003]
发明目的:本发明的目的是开发一种具有原料易得,操作方便,反应易于控制,收率较高的合成2-氟环丁基甲胺及其中间体的方法。
[0004]
本发明提供了一种化合物i的制备方法:
[0005][0006]
其中,化合物iv制备化合物v的步骤中,磺酰化试剂选自甲基磺酰氯、乙基磺酰氯、对甲基苯磺酰氯、甲基磺酸酐或者三氟甲磺酸酐;r1为甲基磺酰基、乙基磺酰基、对甲苯磺酰基或者三氟甲磺酰基;
[0007]
化合物v制备化合物vi的步骤中,胺化试剂选自叠氮化钠、苄胺、二苄胺或者邻苯二甲酰亚胺钾盐;r2为
[0008]
当r2为时,化合物vi制备化合物i的步骤中,需要加入还原剂2;
[0009]
当r2为时;化合物vi制备化合物i的步骤中,需要加入脱保护试剂。
[0010]
优选的,化合物vi制备化合物i的步骤中:
[0011]
当r2为时,还原剂2选自氢气、氢化铝锂或者三苯基膦;当还原剂2为氢气时,需要加入催化剂钯碳或氢氧化钯碳;
[0012]
当r2为时,脱保护试剂选自钯碳/氢气或者氢氧化钯碳/氢气;
[0013]
当r2为时,脱保护试剂选自水合肼、甲胺或者盐酸;
[0014]
优选的,化合物ii制备化合物iii的步骤中,化合物ii、选择性氟试剂和硝酸银的摩尔比范围为1∶1~3∶0.01~0.5;反应温度范围为0~80℃;
[0015]
优选的,化合物iii制备化合物iv的步骤中,反应还原剂1选自硼烷四氢呋喃、硼烷二甲硫醚或者四氢铝锂;
[0016]
优选的,化合物iii制备化合物iv的步骤中,化合物iii与还原剂1的摩尔比范围是1∶0.75~1∶2;
[0017]
优选的,化合物iii制备化合物iv的步骤中,反应温度范围为0~40℃;
[0018]
优选的,化合物iv制备化合物v的步骤中,化合物iv与磺酰化试剂的摩尔比范围为1∶1~1∶2;
[0019]
优选的,化合物iv制备化合物v的步骤中,碱选自三乙胺、二异丙基乙胺、吡啶、氢氧化钠或者氢氧化钾;化合物iv与碱的摩尔比范围为1∶1~1∶5;
[0020]
优选的,化合物iv制备化合物v的步骤中,反应温度范围为0~40℃;
[0021]
优选的,化合物v制备化合物vi的步骤中,化合物v与胺化试剂的摩尔比范围为1∶1~1∶3;反应温度范围为60~120℃。
[0022]
有益效果
[0023]
本发明提供了一种合成2-氟环丁基甲胺及其中间体的方法,该方法合成路线可靠,反应易于放大,操作方便,具有工业化应用前景。
[0024]
说明书中涉及到的反应试剂的缩写如下所示:
[0025]
mscl:甲基磺酰氯;
[0026]
tscl:对甲苯磺酰氯;
[0027]
tea:三乙胺;
[0028]
dipea:n,n-二异丙基乙胺;
[0029]
pe:石油醚;
[0030]
thf:四氢呋喃;
[0031]
mtbe:甲基叔丁基醚;
[0032]
dcm:二氯甲烷;
[0033]
ea:乙酸乙酯。
具体实施方式
[0034]
下面结合具体实施例,进一步阐明本发明,本实施例在以本发明技术方案为前提下进行实施,应理解这些实施例仅用于说明本发明而不用于限制本发明的范围。
[0035]
实施例1
[0036][0037]
化合物iii的制备
[0038]
将化合物ii(40.0g,0.278mol,1.0eq.)溶于800ml丙酮中,加入硝酸银(9.4g,0.0555mol,0.2eq.)、选择性氟试剂(196.6g,0.555mol,2.0eq.)、800ml水、过硫酸钠(33.1g,0.139mol,0.5eq.),25℃搅拌反应24h。减压浓缩除去丙酮,用3*200ml ea萃取,再用3*200ml mtbe萃取,合并有机相,用200ml饱和食盐水洗涤,无水硫酸镁干燥,减压浓缩,得粗品为深黄色液体,制砂柱层析纯化(正庚烷/ea洗脱,得化合物iii为黄色液体20.0g,gc检测纯度95%,收率:60.9%。1h nmr(400mhz,cdcl3)δ(ppm):5.05~5.35(m,1h),3.25~3.53(m,1h),2.20~2.60(m,3h),1.73~1.84(m,1h)。
[0039]
化合物iv的制备
[0040]
将化合物iii(8.0g,0.0677mol,1.0eq.)溶于20ml thf中,冰浴下滴加1.0m硼烷四氢呋喃溶液(81.3ml,0.0813mol,1.2eq.),控温低于20℃,滴毕,20℃搅拌反应2h。gc检测显示原料反应完全。滴加甲醇(6.5g)淬灭反应,减压浓缩反应液,得化合物iv为无色液体7.0g,直接投入下一步反应。
[0041]
化合物v-1的制备
[0042]
将化合物iv(7.00g,0.0672mol,1.0eq.)溶于dcm(100ml)中,加入naoh(5.39g,0.133mol,2.0eq.),tscl(12.81g,0.067mol,1.0eq.),20℃搅拌反应15h,gc检测显示原料反应完全。加入水(80ml),分液,水相用dcm(50ml*2)萃取,饱和食盐水(50ml)洗涤,干燥,浓
缩,得到化合物v-1为无色液体12.39g,两步收率70.9%。
[0043]
化合物vi-1的制备
[0044]
将化合物v-1(12.39g,0.0476mol,1.0eq.)溶于dmf(150ml)中,加入nan3(6.30g,0.0969mol,2.0eq.),加热温度至100℃,搅拌反应3h,tlc检测显示原料反应完全。降温到20℃,加入水(450ml),mtbe(100ml*3)萃取,合并有机相,水(100ml*3)洗涤有机相,饱和食盐水(100ml)洗涤,干燥,浓缩得到化合物vi-1为黄色液体6.15g,直接投入下一步反应。
[0045]
化合物i的制备
[0046]
将化合物vi-1(6.15g,0.0476mol,1.0eq.)溶于thf(80ml)中,加入pph3(24.92g,0.0952mol,2.0eq.),h2o(5.14g,0.286mol,6.0eq),25℃搅拌反应15h,lc-ms检测无过渡态残留,直接处理。用2n hcl调节ph=2,浓缩反应液,dcm(100ml*3)萃取,水相用15%氢氧化钠水溶液调节ph=7,二甲基四氢呋喃萃取,合并有机相,干燥,浓缩制砂柱层析纯化(dcm/meoh洗脱),得化合物i为淡黄色液体2.91g,收率60.5%。1h nmr(400mhz,cdcl3)δ(ppm):3.70~3.72(m,1h),2.44~2.79(m,3h),1.47~1.69(m,2h),1.72~1.94(m,2h)。(esi-tof)m/z:[m+h]
+
calcd for c5h
10
nf:103;found:104。
[0047]
实施例2
[0048][0049]
化合物iii的制备
[0050]
将化合物ii(40.0g,0.278mol,1.0eq.)溶于800ml乙腈中,加入硝酸银(23.61g,0.139mol,0.5eq.)、选择性氟试剂(295.4g,0.834mol,3.0eq.)、800ml水,80℃搅拌反应4h。冷却后,减压浓缩除去乙腈,用3*200ml ea萃取,再用3*200ml mtbe萃取,合并有机相,用200ml饱和食盐水洗涤,无水硫酸镁干燥,减压浓缩,得粗品为深黄色液体,制砂柱层析纯化(正庚烷/ea洗脱,得化合物iii为黄色液体21.44g,gc检测纯度95%,收率:65.3%。1h nmr(400mhz,cdcl3)δ(ppm):5.05~5.35(m,1h),3.25~3.53(m,1h),2.20~2.60(m,3h),1.73~1.84(m,1h)。
[0051]
化合物iv的制备
[0052]
0℃下,将lah(2.41g,0.0634mol,0.75eq.)悬浮于10ml thf中,分批加入化合物iii(9.99g,0.0846mol,1.0eq.),加完升温至室温搅拌反应2h。gc检测显示原料反应完全。往反应液中分别滴加2.41g水,2.41g 15%naoh水溶液,7.23g水,加完搅拌后抽滤,thf淋洗,合并干燥后,减压浓缩反应液,得化合物iv为无色液体9.3g,直接投入下一步反应。
[0053]
化合物v-2的制备
[0054]
将化合物iv(9.00g,0.0846mol,1.0eq.)溶于dcm(100ml)中,加入tea(42.76g,0.423mol,5.0eq.),mscl(19.38g,0.169mol,2.0eq.),20℃搅拌反应15h,gc检测显示原料反应完全。加入水(100ml),分液,水相用dcm(50ml*2)萃取,合并有机相,饱和食盐水(50ml)洗涤,干燥,浓缩,得到化合物v-2为无色液体10.10g,两步收率65.5%。
[0055]
化合物vi-2的制备
[0056]
将化合物v-2(10.01g,0.055mol,1.0eq.)溶于dmf(80ml)中,加入二苄胺(32.52g,0.165mol,3.0eq.),加热升温至120℃,搅拌反应5h,tlc检测显示原料反应完全。降温到20℃,加入水(300ml),mtbe(100ml*3)萃取,合并有机相,水(100ml*3)洗涤有机相,饱和食盐水(100ml)洗涤,干燥,浓缩后制砂柱层析纯化(正庚烷/ea洗脱),得化合物vi-2为黄色固体12.47g,收率80%。
[0057]
化合物i的制备
[0058]
高压釜中,将化合物vi-1(12.47g,0.044mol,1.0eq.)溶于meoh(100ml)中,加入pd(oh)2/c(2g),2mpa氢气氛围下,40℃搅拌反应16h,lc-ms检测原料反应完全,抽滤除去残渣,母液浓缩后浓缩制砂柱层析纯化(dcm/meoh洗脱),得化合物i为淡黄色液体3.20g,收率70.5%。1h nmr(400mhz,cdcl3)δ(ppm):3.70~3.72(m,1h),2.44~2.79(m,3h),1.47~1.69(m,2h),1.72~1.94(m,2h)。(esi-tof)m/z:[m+h]
+
calcd for c5h
10
nf:103;found:104。
[0059]
实施例3
[0060][0061]
化合物iii的制备
[0062]
将化合物ii(40.0g,0.278mol,1.0eq.)溶于800ml丙酮中,0℃下加入硝酸银(0.472g,0.00278mol,0.01eq.)、选择性氟试剂(98.48g,0.278mol,1.0eq.),加完室温搅拌反应10h。冷却后,将反应液倒入1000ml水中,搅拌后mtbe萃取,合并有机相后,干燥,减压浓缩,得粗品为深黄色液体,制砂柱层析纯化(正庚烷/ea洗脱,得化合物iii为黄色液体22.42g,gc检测纯度95%,收率:68.3%。1h nmr(400mhz,cdcl3)δ(ppm):5.05~5.35(m,1h),3.25~3.53(m,1h),2.20~2.60(m,3h),1.73~1.84(m,1h)。
[0063]
化合物iv的制备
[0064]
将化合物iii(8.0g,0.0677mol,1.0eq.)溶于20ml thf中,冰浴下滴加1.0m硼烷二甲硫醚溶液(135ml,0.135mol,2.0eq.),控温低于20℃,滴毕,40℃搅拌反应1h。gc检测显示
原料反应完全。滴加甲醇(6.5g)淬灭反应,减压浓缩反应液,得化合物iv为无色液体8.0g,直接投入下一步反应。
[0065]
化合物v-3的制备
[0066]
将化合物iv(8.00g,0.0677mol,1.0eq.)溶于dcm(100ml)中,加入dipea(8.75g,0.0677mol,1.0eq.),三氟甲磺酸酐(19.10g,0.0677mol,1.0eq.),20℃搅拌反应15h,gc检测显示原料反应完全。加入水(100ml),分液,水相用dcm(50ml*2)萃取,合并有机相,饱和食盐水(50ml)洗涤,干燥,浓缩,得到化合物v-3为无色液体10.15g,两步收率63.5%。
[0067]
化合物vi-3的制备
[0068]
将化合物v-3(10.01g,0.0423mol,1.0eq.)溶于dmf(100ml)中,加入苄胺(4.54g,0.0423mol,1.0eq.),加热升温至80℃,搅拌反应15h,tlc检测显示原料反应完全。降温到20℃,加入水(300ml),mtbe(100ml*3)萃取,合并有机相,水(100ml*3)洗涤有机相,饱和食盐水(100ml)洗涤,干燥,浓缩后制砂柱层析纯化(正庚烷/ea洗脱),得化合物vi-3为黄色固体5.60g,收率68.5%。
[0069]
化合物i的制备
[0070]
高压釜中,将化合物vi-3(5.01g,0.0259mol,1.0eq.)溶于meoh(50ml)中,加入pd/c(1g),3mpa氢气氛围下,50℃搅拌反应16h,lc-ms检测原料反应完全,抽滤除去残渣,母液浓缩后浓缩制砂柱层析纯化(dcm/meoh洗脱),得化合物i为淡黄色液体2.02g,收率75.5%。1h nmr(400mhz,cdcl3)δ(ppm):3.70~3.72(m,1h),2.44~2.79(m,3h),1.47~1.69(m,2h),1.72~1.94(m,2h)。(esi-tof)m/z:[m+h]
+
calcd for c5h
10
nf:103;found:104。
[0071]
实施例4
[0072][0073]
化合物vi-4的制备将化合物v-1(50.01g,0.193mol,1.0eq.)(参考实施例1-3的方法制备化合物v-1)溶于dmf(500ml)中,加入ki(50.0g),邻苯二甲酰亚胺钾盐(53.62g,0.289mol,1.5eq.)加热温度至60℃,搅拌反应8h,tlc检测显示原料反应完全。降温到20℃,加入水(1200ml),搅拌30min后,抽滤,得褐色固体,溶解后制砂柱层析纯化(正庚烷/ea洗脱),得化合物vi-4为咖啡色固体29.49g,收率65.5%。(esi-tof)m/z:[m+h]
+
calcd for c
13
h
12
no2f:233;found:234。
[0074]
化合物i的制备
[0075]
将化合物vi-4(25.01g,0.107mol,1.0eq.)溶于meoh(300ml)中,加入水合肼(10.73g,0.214mol,2.0eq.),加热至回流搅拌反应15h,lc-ms检测显示原料反应完全,将反应液倒至1l水中,甲基四氢呋喃萃取,合并有机相,干燥后浓缩制砂柱层析纯化(dcm/meoh洗脱),得化合物i为淡黄色液体7.23g,收率65.5%。1h nmr(400mhz,cdcl3)δ(ppm):3.70~3.72(m,1h),2.44~2.79(m,3h),1.47~1.69(m,2h),1.72~1.94(m,2h)。(esi-tof)m/z:[m
+h]
+
calcd for c5h
10
nf:103;found:104。
[0076]
实施例5
[0077][0078]
化合物vi-1的制备将化合物v-2(10.01g,0.0549mol,1.0eq.)(参考实施例1-3的方法制备化合物v-2)溶于dmf(100ml)中,加入nan3(5.36g,0.0824mol,1.5eq.),加热温度至60℃,搅拌反应5h,tlc检测显示原料反应完全。降温到20℃,加入水(450ml),mtbe(100ml*3)萃取,合并有机相,水(100ml*3)洗涤有机相,饱和食盐水(100ml)洗涤,干燥,浓缩得到化合物vi-1为黄色液体7.21g,直接投入下一步反应。
[0079]
化合物i的制备
[0080]
高压釜中,将化合物vi-1(7.21g,0.0549mol,1.0eq.)溶于meoh(50ml)中,加入pd(oh)2/c(1g),2.5mpa氢气氛围下,60℃搅拌反应16h,lc-ms检测原料反应完全,抽滤除去残渣,母液浓缩后浓缩制砂柱层析纯化(dcm/meoh洗脱),得化合物i为淡黄色液体3.71g,收率65.5%。1h nmr(400mhz,cdcl3)δ(ppm):3.70~3.72(m,1h),2.44~2.79(m,3h),1.47~1.69(m,2h),1.72~1.94(m,2h)。(esi-tof)m/z:[m+h]
+
calcd for c5h
10
nf:103;found:104。
[0081]
实施例6
[0082][0083]
化合物vi-4的制备将化合物v-2(50.01g,0.274mol,1.0eq.)(参考实施例1-3的方法制备化合物v-2)溶于dmf(500ml)中,加入ki(50.0g),邻苯二甲酰亚胺钾盐(76.24g,0.412mol,1.5eq.)加热温度至80℃,搅拌反应6h,tlc检测显示原料反应完全。降温到20℃,加入水(1200ml),搅拌30min后,抽滤,得褐色固体,溶解后制砂柱层析纯化(正庚烷/ea洗脱),得化合物vi-4为咖啡色固体45.05g,收率70.5%。(esi-tof)m/z:[m+h]
+
calcd for c
13
h
12
no2f:233;found:234。
[0084]
化合物i的制备
[0085]
将化合物vi-4(20.51g,0.0879mol,1.0eq.)溶于33%的甲胺甲醇溶液(150ml)中,25℃搅拌反应16h,tlc检测显示原料反应完全,将反应液浓缩干,加入200ml石油醚打浆,过滤,滤液浓缩制砂柱层析纯化(dcm/meoh洗脱),得化合物i为淡黄色液体4.86g,收率53.6%。1h nmr(400mhz,cdcl3)δ(ppm):3.70~3.72(m,1h),2.44~2.79(m,3h),1.47~1.69(m,2h),1.72~1.94(m,2h)。(esi-tof)m/z:[m+h]
+
calcd for c5h
10
nf:103;found:104。
[0086]
实施例6
[0087][0088]
化合物i的制备将化合物vi-4(参考实施例1-5的方法制备化合物vi-4))(15.00g,0.0643mol,1.0eq.)加入到8mol/l的100ml盐酸水溶液中,加热到100℃,搅拌反应16h。tlc检测显示原料反应完全,过滤,滤液用3*100ml ea萃取杂质,水相旋干,再用50ml水溶解,用3*80ml dcm萃取,无水硫酸钠干燥,过滤,滤液旋干得到得化合物i为淡黄色液体5.0g,收率75.4%。1h nmr(400mhz,cdcl3)δ(ppm):3.70~3.72(m,1h),2.44~2.79(m,3h),1.47~1.69(m,2h),1.72~1.94(m,2h)。(esi-tof)m/z:[m+h]
+
calcd for c5h
10
nf:103;found:104。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签:

 热门咨询
热门咨询




tips