
 商标分类
商标分类  商标转让
商标转让 
一种芳香胺取代的斑蝥素衍生物及其制备方法、其药物组合物及其应用与流程
 2021-02-01 22:02:42|
2021-02-01 22:02:42| 377|
377| 起点商标网
起点商标网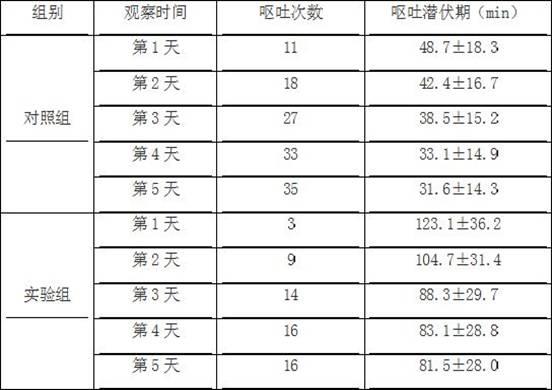
[0001]
本发明属于药物技术领域,具体涉及一种芳香胺取代的斑蝥素衍生物及其制备方法,以其为活性成分的药物组合物,以及它们在制备防治抗肿瘤、抗病毒药物和功能食品中的应用。
背景技术:
[0002]
斑蝥素(1,2-二甲基-3,6-氧代环己烷-1,2-邻二甲酸酐, 1,2-dimethyl-3,6-epoxycyclohexane-1,2-dicarboxylic anhydride)是从昆虫纲动物斑蝥体液中分离得到具有亲酯性的萜类化合物。在室温下斑蝥素是一种无色无味的晶状固体。医学上斑蝥素外用具有皮肤止痒、改善局部神经营养及刺激毛根,促进毛发生长的作用,已经被用于治疗各种皮肤病(痔疮、溃疡、寻常疣和软疣等)。同时临床研究显示斑蝥素是一种丝氨酸/苏氨酸蛋白磷酸酶pp1和pp2a的抑制剂,可导致多种细胞效应,如dna损伤,细胞周期停滞和细胞凋亡等,因此能够显著的抑制肝癌,胰腺癌,结肠癌,肺癌,黑色素瘤和膀胱癌细胞的生长。然而斑蝥素严重的肾毒性限制了其作为小分子药物的应用。虽然在近几十年里人们进行了大量的生理和生化研究,希望找到由斑蝥素引起的生长抑制及细胞死亡的原因,但是其主要的作用和通路分子机制仍然还不清楚。
近年来,人们将斑蝥素的多种衍生物作为潜在的抗肿瘤剂进行了深入研究,如申请号为14/504,564的美国专利公开了斑蝥素及其衍生物在制备抗肿瘤药物中的应用;申请号为10/703,336的美国专利公开了改进的斑蝥酸酐衍生物在抗肿瘤中的应用;申请号为cn201610451738.9的中国专利公开了一种去甲斑蝥素单酸单酯衍生物及其抗肿瘤应用。
[0003]
临床上,这些衍生物具有与斑蝥素一样可引起细胞生长抑制活性的能力,然而也具有强抑制的肾毒性和肠胃毒性。斑蝥酸代表了最早商业化的斑蝥素抗肿瘤剂。但是,这些化合物也受限于它们对正常细胞的毒性,阻止了其作为有效的抗癌药物或化学疗法用于癌症的治疗。
[0004]
同时,在申请号为cn01140066.8的中国专利中,公开了斑蝥酸酐的药物组合物具有抗病毒的作用,在申请号为cn200710087156.8的中国专利中,公开了去甲斑蝥素及其衍生物在预防和治疗艾滋病中的新用途,然而,仍然由于这类化合物具有较强的肾毒性和肠胃毒性,而限制了其使用。
[0005]
因此,迫切需要开发其他新的斑蝥素衍生物来解决这些问题。
技术实现要素:
[0006]
本发明的目的,在于提供化合物芳香胺取代的斑蝥素衍生物为有效成分的抗肿瘤、抗病毒药物及其制备方法,及其此活性成分在制备抗肿瘤、抗病毒药物和制备抗肿瘤功能食品中的应用。
[0007]
本发明的目的,是通过以下技术方案实现的:一种斑蝥素衍生物,具有以下式(i)所示的结构通式:
(i)其中,x和y分别选自:氮,氧,碳,羰基;r1选自:芳基、芳香杂环基及生物活性分子。
[0008]
优选的,式(i)中的斑蝥素衍生物,其中,x优选为氮;y优选为羰基,r1优选为芳基、芳香杂环基。
[0009]
更优选的方案中,上述斑蝥素衍生物为化合物8、化合物9、化合物13、化合物14、化合物15、化合物16、化合物17、化合物18、化合物19、化合物20、化合物29、化合物30,化合物30,化合物30,化合物30,化合物30,化合物30,化合物30,化合物30,化合物30,化合物30,化合物30,化合物30,。
[0010]
本发明还提供了上述斑蝥素衍生物的制备方法,为通过在冰醋酸溶液中缩合芳香胺与斑蝥素。
[0011]
优选地,芳香胺与斑蝥素的缩合反应温度为115-145
°
c,反应时间为12-36h,更优选的,反应温度为135
°
c,反应时间为12h。
[0012]
本发明还提供了一种药物组合物,含有式(i)所示的斑蝥素衍生物、其药学上可接受的盐、溶剂合物及可药用载体或赋形剂中的一种或多种。
[0013]
同时还提供了另一种药物组合物,含有化合物8、化合物9、化合物13、化合物14、化合物15、化合物16、化合物17、化合物18、化合物19、化合物20、化合物29、化合物30及其药学上可接受的盐、溶剂合物及可药用载体或赋形剂中的一种或多种。
[0014]
本发明提供的式(i)斑蝥素衍生物、其药学上可接受的盐、溶剂合物在制备抗肿瘤、抗病毒药物中的应用。
[0015]
本发明提供的式(i)斑蝥素衍生物、其药学上可接受的盐、溶剂合物在制备功能食品中的应用。
[0016]
化合物8、化合物9、化合物13、化合物14、化合物15、化合物16、化合物17、化合物18、化合物19、化合物20、化合物29、化合物30及其药学上可接受的盐、溶剂合物在制备功能食品中的应用。
[0017]
其中,本说明书所用的术语“芳基,芳香杂环基”指单环,稠环芳香烃基,芳香杂环烃基,优选碳原子数为3-14的芳香烃基,芳香杂环烃基,包括苯基,萘基, 蒽基,菲基,苊基,吡咯基,唑基,咪唑基,噻唑基,咔唑基、吡啶基,吗啉基,哌嗪基,吡嗪基,吡唑基,吲哚基,喹啉基等;芳香杂环烃基,包括苯基,吡咯基,吡啶基,吗啉基,哌嗪基,吲哚基等。芳香基,芳香杂环基的取代基团有;氢,烷基,烯基、炔基、卤素,卤素取代烷基,烷氧基,卤素取代烷氧基,烷氨基,卤素取代烷氨基,硝基,氰基,烷氧羰基,烷氧烷氧基,烷氧烷氧基。
[0018]
术语“芳香胺”指氨基取代的芳香基或芳香杂环基。
[0019]
术语“药物组合物”指包含指定量的各指定成分的产品,以及直接或间接从指定量的各指定成分的组合产生的任何产品。
[0020]
术语“药学上可接受的盐”指衍生自无机或有机酸或碱的任何盐。
[0021]
本发明的式(i)化合物用作药物时,可以直接使用,或者以药物组合物的形式使用。该药物组合物含有0.1-99%,优选为0.5-90%的本发明的式(i)化合物, 其余为药物上可接受的,对人体和动物无毒和惰性的可药用载体和/或赋形剂。
[0022]
所述的药用载体或赋形剂是一种或多种选自固体、半固体和液体稀释剂、超填料以及药物制品辅剂。将本发明的药物组合物以单位体重服用量的形式使用。斑蝥素衍生物的组合物采用制药和食品领域公认的方法制备成各种剂型,如液体制剂(注射剂、混悬剂、乳剂、 溶液剂、糖浆剂等)、固体制剂(片剂、胶囊剂、颗粒剂、冲剂等) 、喷剂、气雾剂等。本发明的药物可经注射(静脉注射、静脉滴注、 肌肉注射、腹腔注射、皮下注射)和口服、舌下给药、粘膜透析等给药途径进行抗肿瘤、抗病毒的治疗。
[0023]
附图说明
[0024]
图1:实施组对小鼠血清中bun水平的影响;图2:光学显微镜下肾组织块的病理生理变化。
[0025]
具体实施方式
[0026]
下面结合本发明的实施例对本发明进行进一步的解释说明,但本发明的内容并不局限与此。
[0027]
实施例1化合物8的制备
在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐 (39.2 mg, 0.2 mmol)、2-氨基-3-甲基吡啶 (26.0 mg, 0.24mmol)和冰醋酸 (0.5 ml),用聚四氟乙烯胶塞密封,油浴加热至135
°
c,搅拌反应12小时。反应结束后,将混合液冷却到室温,减压浓缩,得到白色固体8 (16.0-34 mg, 28-59% yield)。1h nmr (300 mhz, cdcl3) δ 8.46 (d, j = 3.8 hz, 1h), 7.68 (d, j = 8.3 hz, 1h), 7.32
ꢀ–ꢀ
7.28 (m, 1h), 4.72
ꢀ–ꢀ
4.71 (m, 2h), 2.25 (s, 3h), 1.90
ꢀ–ꢀ
1.82 (m, 2h), 1.77
ꢀ–ꢀ
1.74 (m, 2h), 1.28 (s, 6h) ppm;ms (esi)m/z calcd for [c
16
h
18
n2o3+na]
+ 309.3, found 309.1;ms (esi)m/z calcd for [c
16
h
18
n2o3+h]
+ 287.1390, found 287.1386。
[0028]
化合物9的制备在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐 (39.2 mg, 0.2 mmol)、4-氨基二苯醚 (44.5 mg, 0.24 mmol) 和冰醋酸 (0.5 ml),用聚四氟乙烯胶塞密封,油浴加热至135
ꢀ°
c,搅拌反应12小时。反应结束后,将混合液冷却到室温,减压浓缩,得到淡黄色固体9 (19.0-36.0 mg, 29-50% yield)。1h nmr (300 mhz, cdcl3) δ 7.37
ꢀ–ꢀ
7.33 (m, 2h), 7.27
ꢀ–ꢀ
7.24 (m, 2h), 7.15
ꢀ–ꢀ
7.11 (m, 1h), 7.06
ꢀ–ꢀ
7.03 (m, 4h), 4.68 (s, 2h), 1.87
ꢀ–ꢀ
1.84 (m, 2h), 1.75
ꢀ–ꢀ
1.73 (m, 2h), 1.24 (s, 6h) ppm;ms (esi)m/z calcd for [c
22
h
21
no4+na]
+ 386.4, found 386.2;ms (esi)m/z calcd for [c
22
h
21
no4+h]
+ 364.1543, found 364.1540。
[0029]
化合物13的制备在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐 (98.1 mg, 0.5 mmol)、6-氨基苯并噻唑 (90.1 mg, 0.6 mmol) 和冰醋酸 (1.0 ml),用聚四氟乙烯胶塞密封,油浴加热至135
ꢀ°
c,搅拌反应12小时。反应结束后,将混合液冷却到室温,减压浓缩,粗品通过薄层
层析硅胶板分离(石油醚:乙酸乙酯=3:1),得到白色固体13 (13.5
ꢀ-
45.0mg)。1h nmr (300 mhz, cdcl3) δ 9.05 (s, 1h), 8.20 (d, j = 8.7 hz, 1h), 7.95 (s, 1h), 7.46 (d, j = 8.7 hz, 1h), 4.72 (s, 2h), 1.89
ꢀ–ꢀ
1.75 (m, 4h), 1.28 (s, 6h) ppm;
13
c nmr (75 mhz, cdcl3) δ 180.6, 155.4, 152.9, 134.1, 129.5, 124.8, 123.9, 120.3, 84.1, 54.1, 23.8, 12.9 ppm;hrms (esi)m/z calcd for [c
17
h
16
n2o3s+h]
+ 329.0954, found 329.0957。
[0030]
化合物14的制备在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐 (98.1 mg, 0.5 mmol)、5-氨基茚旦 (79.14 mg, 0.6 mmol) 和冰醋酸 (1.0 ml),用聚四氟乙烯胶塞密封,油浴加热至135
ꢀ°
c,搅拌反应12小时。反应结束后,将混合液冷却到室温,减压浓缩,粗品通过薄层层析硅胶板分离(石油醚:乙酸乙酯=3:1),得到白色固体14 (81.3-132 mg)。1h nmr (300 mhz, cdcl3) δ 7.28 (d, j = 7.3 hz, 1h), 7.11 (s, 1h), 7.00 (d, j = 7.6 hz, 1h), 4.68 (m, j = 1.7 hz, 2h), 2.91 (d, j = 5.9 hz, 4h), 2.13
ꢀ–ꢀ
2.03 (m, 2h), 1.86
ꢀ–ꢀ
2.03 (m, 4h), 1.23 (s, 6h) ppm;
13
c nmr (75 mhz, cdcl3) δ 180.8, 145.2, 144.8, 129.9, 124.6, 124.3, 122.5, 83.9, 53.9, 32.7, 32.5, 25.4, 23.7, 12.7 ppm;hrms (esi)m/z calcd for [c
19
h
21
no3+h]
+ 312.1594, found 312.1593。
[0031]
化合物15的制备在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐 (98.1 mg, 0.5 mmol)、对甲氧基苯胺 (74.0 mg, 0.6mmol)和冰醋酸 (1.0 ml),用聚四氟乙烯胶塞密封,油浴加热至135
°
c,搅拌反应12小时。反应结束后,将混合液冷却到室温,减压浓缩,粗品通过薄层层析硅胶板分离(石油醚:乙酸乙酯=3:1),得到白色固体15 (28.7
ꢀ-
79.0mg)。1h nmr (300 mhz, cdcl3) δ 7.20 (d, j = 8.9 hz, 2h), 6.96 (d, j = 8.9 hz, 2h), 4.68 (s, 2h), 3.81 (s, 3h), 1.89
ꢀ–ꢀ
1.71 (m, 4h), 1.24 (s, 6h) ppm;
13
c nmr (75 mhz, cdcl3) δ 180.8, 159.3, 127.6, 124.6, 114.2, 83.9, 55.4, 53.9, 23.7, 12.8 ppm;hrms (esi)m/z calcd for [c
17
h
19
no4+h]
+ 302.1387, found 302.1385。
[0032]
化合物16的制备
在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐 (98.1 mg, 0.5 mmol)、4-乙炔基苯胺 (64.5 mg, 0.55 mmol) 和冰醋酸 (1.0 ml),用聚四氟乙烯胶塞密封,油浴加热至135
ꢀ°
c,搅拌反应12小时。反应结束后,将混合液冷却到室温,减压浓缩,粗品通过薄层层析硅胶板分离(石油醚:乙酸乙酯=3:1),得到白色固体16 (10.0-35 mg)。1h nmr (300 mhz, cdcl3) δ 8.05 (d, j = 8.2 hz, 2h), 7.46 (d, j = 8.3 hz, 2h), 4.71 (s, 2h), 2.62 (s, 3h), 1.89
ꢀ–ꢀ
1.75 (m, 4h), 1.27 (s, 6h) ppm;
13
c nmr (75 mhz, cdcl3) δ 197.0, 180.2, 136.6, 136.1, 129.0, 126.4, 84.1, 54.2, 26.7, 23.8, 12.9 ppm;hrms (esi)m/z calcd for [c
18
h
19
no4+h]
+ 314.1387, found 314.1385。
[0033]
化合物17的制备在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐(98.1 mg, 0.5 mmol)、3-氨基-9-乙基咔唑 (126.9 mg, 0.6 mmol) 和冰醋酸 (1.0 ml),用聚四氟乙烯胶塞密封,油浴加热至135
ꢀ°
c,搅拌反应12小时。反应结束后,将混合液冷却到室温,减压浓缩,粗品通过薄层层析硅胶板分离(石油醚:乙酸乙酯=3:1),得到白色固体17 (28.1
ꢀ-
79mg)。1h nmr (300 mhz, cdcl3) δ 8.06 (d, j = 7.7 hz, 1h), 7.99 (s, 1h), 7.50
ꢀ–ꢀ
7.38 (m, 3h), 7.32 (dd, j = 8.6 hz, 1h), 7.25
ꢀ–ꢀ
7.20 (m, 1h), 4.75 (s, 2h), 4.38
ꢀ–ꢀ
4.31 (m, 2h), 1.88
ꢀ–ꢀ
1.74 (m, 4h), 1.40 (t, j = 7.1 hz, 3h), 1.28 (s, 6h) ppm;
13
c nmr (75 mhz, cdcl3) δ 181.3, 140.3, 139.4, 126.0, 123.9, 123.1, 122.5, 120.7, 119.0, 108.7, 108.6, 84.0, 53.9, 37.6, 23.8, 13.7, 12.9 ppm;hrms (esi)m/z calcd for [c
24
h
24
n2o3+h]
+ 389.1860, found 389.1856。
[0034]
化合物18的制备
125.6, 124.0, 84.1, 54.1, 23.8, 12.9 ppm;hrms (esi)m/z calcd for [c
20
h
19
no3+h]
+ 322.1438, found 322.1436。
[0037]
化合物29的制备在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐 (98.1 mg, 0.5 mmol)、4-异丙基苯胺 (75 μl, 0.6 mmol) 和冰醋酸 (2.0 ml),用聚四氟乙烯胶塞密封,油浴加热至135
ꢀ°
c,搅拌反应12小时。反应结束后,将混合液冷却到室温,减压浓缩,粗品通过薄层层析硅胶板分离(石油醚:乙酸乙酯=3:1),得到白色固体29 (60.0-97.0 mg, 34-79% yield)。1h nmr (300 mhz, cdcl3) δ 7.32
ꢀ–ꢀ
7.19 (m, 4h), 4.69 (s, 2h), 2.95
ꢀ–ꢀ
2.91 (m, 1h), 1.84
ꢀ–ꢀ
1.75 (m, 4h), 1.26
ꢀ–ꢀ
1.23 (m, 12h) ppm;
13
c nmr (75 mhz, cdcl3) δ 180.8, 149.3, 129.6, 127.1, 126.3, 84.1, 54.0, 33.9, 23.9, 23.8, 12.9 ppm;ms (esi)m/z calcd for [c
19
h
23
no3+h]
+
314.1751, found 314.1755。
[0038]
化合物30的制备在装有搅拌子的10毫升耐压反应管中依次加入斑蝥酸酐 (98.1 mg, 0.5 mmol)、叔丁基苯胺 (88 μl, 0.6 mmol) 和冰醋酸 (2.0 ml),用聚四氟乙烯胶塞密封,油浴加热至115-145
ꢀ°
c,搅拌反应12-36小时。反应结束后,将混合液冷却到室温,减压浓缩,粗品通过薄层层析硅胶板分离(石油醚:乙酸乙酯 = 3:1),得到白色固体30 (52.7-97 mg, 24-89% yield)。1h nmr (300 mhz, cdcl3) δ 7.47 (d, j = 8.1 hz, 2h), 7.22 (d, j = 8.2 hz, 2h), 4.69 (s, 2h), 1.87
ꢀ–ꢀ
1.67 (m, 4h), 1.32
ꢀ–ꢀ
1.25 (m, 15h) ppm;
13
c nmr (75 mhz, cdcl3) δ 180.8, 151.5, 129.3, 126.1, 125.9, 84.1, 54.0, 31.3, 23.8, 23.4, 12.9 ppm;ms (esi)m/z calcd for [c
20
h
25
no3+h]
+
328.1907, found 328.1904。
[0039]
实施例2:按实施例1的方法先制得斑蝥素衍生物,按常规加注射液用水,精滤,灌封灭菌制成注射液。
[0040]
实施例3:按实施例1的方法先制得斑蝥素衍生物,将其溶于无菌注射用水中,搅拌使溶,用无菌抽滤漏斗过滤,再无菌精滤,分装于安瓿中,低温冷冻干燥后灭菌熔封得粉针剂。
[0041]
实施例4:按实施例1的方法先制得斑蝥素衍生物,按其与赋形剂重量比为 9:1 的比例加入赋形
剂,制成粉剂。
[0042]
实施例5:按实施例1的方法先制得斑蝥素衍生物,按其与赋形剂重量比为 1:5-1:10的比例加入赋形剂,制粒压片。
[0043]
实施例6:按实施例1的方法先制得斑蝥素衍生物,按常规口服液制法制成口服液。
[0044]
实施例7:按实施例1的方法先制得斑蝥素衍生物,按其与赋形剂重量比为5:1的比例加入赋形剂,制成胶囊或颗粒剂或冲剂。
[0045]
实施例8:按实施例1的方法先制得斑蝥素衍生物,按其与赋形剂重量比为3:1的比例加入赋形剂,制成胶囊或颗粒剂或冲剂。
[0046]
实施例9:取按实施例1的方法先制得斑蝥素衍生物43.6克,加入淀粉570 克,乳糖180克,薄荷醇3克,羧甲基淀粉钠152克,制成含片,作为功能食品。
[0047]
为了更好的理解本发明的实质,下面以本发明的试验例即本发明的式 (i)化合物斑蝥素衍生物与药用载体或赋形剂组成的药物组合物的药理作用结果来说明本发明的优异性,但不以此来限制本发明。
[0048]
试验例1:1.1斑蝥素衍生物(由本发明的上述实施例1所制得)的肾毒性测试动物(雄性icr小鼠,体重20
±
2g,由南京医学院提供)到达实验室后,预饲养一周,给以标准食物,自由饮水。标准条件下适应性饲养后,均分为3组,一组作为空白对照组,腹腔注射给予0.9%生理盐水;一组作为实验组,腹腔注射给以等体积的药物(以本发明的上述实施例1所制得的化合物15为代表);另外一组作为阳性对照组,腹腔注射给以等体积的去甲斑蝥素。实验组和阳性对照组连续给药6天,空白对照组给以等体积的生理盐水。末次给药24小时后,通过摘眼球收集血液,测试肾功能指标,然后,将小鼠通过脱臼处死后,收取肾脏,对肾组织进行病理学分析。
[0049]
1.2血清肾功能指标测试将上述收集到的血液样品在室温下放置2小时后,以3500rpm离心10分钟,收集上层血清。根据试剂盒说明书,用比色测定试剂盒确定血尿素氮(bun)的血清水平(bun是肾细胞损伤的敏感标志物,其血清升高表示肾损伤的发生)。结果如图1所示。
[0050]
由图1可以看出,与空白对照组相比,腹腔注射给予去甲斑蝥素的阳性对照组显著升高血清bun水平,表示去甲斑蝥素能导致肾损伤;采用实验组对血清bun水平稍微升高,表明式(i)所制得的斑蝥衍生物导致肾损伤程度较小。
[0051]
1.3肾组织的病理学观察将肾组织切成5μm的小块,在15%福尔马林中浸泡固定8~10小时,并依次用30%、70%、85%、95%、99.8%(即为无水乙醇)的酒精脱水,每次脱水1小时。在无水乙醇与二甲苯体积比为1:1混合溶液中透明2小时,再用纯二甲苯分别浸渍处理2小时。选取熔点在52~54℃的石蜡,将肾组织块放在熔化的石蜡和二甲苯的体积比为1:1混合溶液中浸泡1~2小时,
再移入熔化的石蜡液中,于55~57℃左右的温箱中浸渍3小时两次,以便于石蜡浸入组织内。将浸蜡后的块状肾组织包埋在石蜡中,并切片处理。制好的切片在37℃恒温干燥箱中干燥,二甲苯脱蜡,逐级经分级浓度的乙醇直至30%酒精,用苏木精-伊红染色法(haematoxylin and eosin,h&e)染色。经苏木精-伊红染色后,苏木精将细胞核染成蓝紫色,其它成分则被伊红染成粉红色。染色后的切片经梯度酒精脱水以及二甲苯透明后,迅速擦去肾组织块周围多余液体,滴加1~2滴中性树胶,将洁净盖玻片倾斜放下(避免出现气泡),封片后就制成永久性的玻片标本,在光学显微镜下观察肾组织块的病理生理变化。结果见图2。
[0052]
如图2所示,空白对照组肾脏结构正常,边界清晰(图2a);图2b为阳性对照组,肾小球肿胀,肾小管坏死,出现空泡,表明去甲斑蝥素会导致小鼠肾脏病理性损伤;而在实验组(图2c)中,肾脏结构趋于正常,表明式(i)所制得的斑蝥衍生物具有较小的肾毒性。
[0053]
试验例2:斑蝥酸衍生物致家鸽的呕吐试验:试验方法:健康成年家鸽(雌雄兼用,450~500g,由南京市江宁区青龙山动物繁殖场提供)到达实验室后,预饲养一周,给以标准食物,自由饮水。标准条件下适应性饲养后,均分为2组,每组20只,其中一组作为实验组,另外一组作为对照组,两组之间无显著性差异,具有可比性,末次给药前禁食12h。所有家鸽接受一个周期(5天)的药物注射,记录家鸽每天出现第一次呕吐的时间(呕吐潜伏期)和每天的呕吐次数。
[0054]
实验组:翼下静脉注射给以本发明药物(4mmol/kg,由本发明的上述实施例1所制得);对照组:翼下静脉注射给以等摩尔的去甲斑蝥素。
[0055]
结果:家鸽呕吐试验结果如下表1所示。
[0056]
表1.本发明的斑蝥衍生物所致的家鸽呕吐试验结果
由表2可知,与对照组相比,本发明的斑蝥素衍生物与对照组相比能明显减少家鸽的呕吐次数,具有较小的胃肠道反应率。
[0057]
试验例3:斑蝥素衍生物(由本发明的上述实施例1所制得)的体外抗肿瘤试验:肿瘤细胞株:肝癌、肺癌、胃癌、胰腺癌、淋巴癌、白血病细胞、宫颈癌瘤株,由中国药科大学药理实验室提供;试验方法:噻唑蓝还原法(mtt)对本发明制备得到的式(i)化合物即斑蝥素衍生物进行体外抗肿瘤试验,分别将肝癌、肺癌、胃癌、胰腺癌、淋巴癌、白血病细胞、宫颈癌瘤株接种到96孔板中,制成浓度为5
×
104个/ml的细胞悬液。向96孔板中,每孔加入100μl细胞悬液(每孔5
×
103个细胞),于37℃、5%co2培养箱中培养24小时;然后,药物用含3%fbs的dmem培养基进行倍比稀释至所需的浓度,给药容积为100μl/孔,同时设立生理盐水空白对照组和去甲斑蝥素、斑蝥酸钠阳性对照组,空白对照组和阳性对照组也分别采用含3%fbs的dmem培养基进行倍比稀释,再将96孔板置于37℃、5%co2培养箱中培养72小时,然后每孔加20μlmtt(5mg/ml,pbs配),继续培养4小时,吸弃上清,每孔加入150μldmso溶解,在酶标仪上,振动10-15分钟轻轻混匀。在λ=570nm的波长下,用酶标仪检测每孔的吸光度即od值,独立实验重复三次以上,以各复孔的平均值作为该细胞的od值,按照下列公式计算生长抑制率(inhibitionratio,ir)。
[0058]
具体试验结果如表2所示:表2斑蝥素衍生物对肿瘤细胞的抑制作用(ic
50
为半数抑制率)
由表2体外抗肿瘤试验结果可知,本发明提供的式(i)化合物即斑蝥素衍生物对多种肿瘤细胞均有较强的抑制作用,可以用于恶性肿瘤疾病的治疗。
[0059]
试验例4:斑蝥素衍生物(由本发明的上述实施例1所制得)的抗病毒试验:病毒细胞:hepg2.2.15细胞、hiv感染的mt4细胞,由中国药科大学药理实验室提供;试验方法,基本同试验例3,不同之处在于采用显微镜观察cpe(病变),设细胞对照组,实验组,阳性对照组1(恩替卡韦),阳性对照组2(齐多夫定),同时观察cpe。实验结果如表3 所示。
[0060]
具体试验结果如表3所示:表3斑蝥素衍生物体外对病毒细胞的抑制作用由表3体外抗病毒试验结果可知,本发明提供的式(i)化合物即斑蝥素衍生物对多种病毒细胞均有抑制作用,可以用于抗病毒疾病的治疗。
[0061]
以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,本领域普通技术人员对本发明的技术方案所做的其他修改或者等同替换,只要不脱离本发明技术方案的精神和范围,均应涵盖在本发明的权利要求范围当中。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips