
 商标分类
商标分类  商标转让
商标转让 
蛋白质的制造方法和二糖的制造方法与流程
 2021-02-01 22:02:08|
2021-02-01 22:02:08| 209|
209| 起点商标网
起点商标网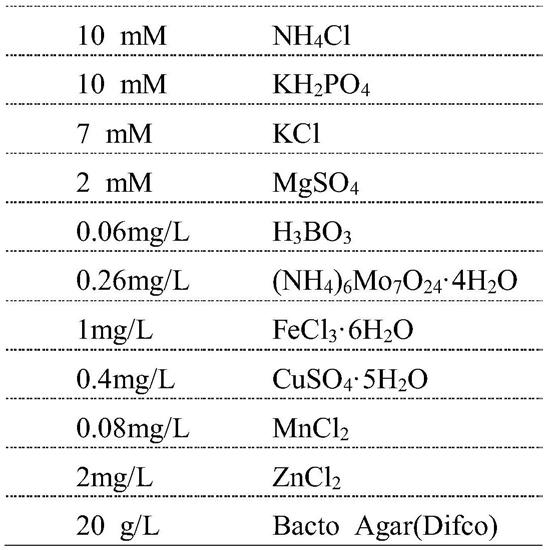
cellulolyticus,其中,
[0038]
所述talaromyces cellulolyticus具有选自下述性质(a)~(c)中的任一种性质:
[0039]
(a)其经过了修饰,使其与非修饰菌株相比gh1-2蛋白的活性降低;
[0040]
(b)其经过了修改,使得gh1-2基因具有提高talaromyces cellulolyticus的生产靶蛋白的能力的变异;
[0041]
(c)它们的组合,
[0042]
所述表达诱导物质为包含葡萄糖作为构成糖的糖,
[0043]
其中,在所述gh1-2蛋白的活性完全消失的情况下,所述表达诱导物质为龙胆二糖。
[0044]
4、所述方法,其中,
[0045]
所述表达诱导物质为龙胆二糖、纤维二糖或纤维素。
[0046]
5、所述方法,其中,
[0047]
所述表达诱导物质为龙胆二糖。
[0048]
6、所述方法,其中,
[0049]
通过选自下述手段(a1)~(a4)中的任一种手段而使所述gh1-2蛋白的活性降低:
[0050]
(a1)降低gh1-2基因的表达;
[0051]
(a2)破坏gh1-2基因;
[0052]
(a3)对gh1-2基因进行修饰使之具有提高talaromyces cellulolyticus的生产靶蛋白的能力的变异;以及
[0053]
(a4)它们的组合。
[0054]
7、所述方法,其中,
[0055]
所述gh1-2蛋白的活性因gh1-2基因的缺失而降低。
[0056]
8、所述方法,其中,
[0057]
所述变异为选自下述变异(a)~(d)中的任一种变异:
[0058]
(a)与seq id no.23所示的氨基酸序列的267位的半胱氨酸残基相当的氨基酸残基被其他氨基酸残基所取代的变异;
[0059]
(b)与seq id no.23所示的氨基酸序列的363位的色氨酸残基相当的氨基酸残基被其他氨基酸残基所取代的变异;
[0060]
(c)与seq id no.23所示的氨基酸序列的449位的色氨酸残基相当的氨基酸残基被其他氨基酸残基所取代的变异;
[0061]
(d)它们的组合。
[0062]
9、所述方法,其中,
[0063]
所述变异(a)~(c)分别为下述变异(a1)~(c1):
[0064]
(a1)与seq id no.23所示的氨基酸序列的267位的半胱氨酸残基相当的氨基酸残基被脯氨酸残基所取代的变异;
[0065]
(b1)与seq id no.23所示的氨基酸序列的363位的色氨酸残基相当的氨基酸残基被苯丙氨酸残基所取代的变异;
[0066]
(c1)与seq id no.23所示的氨基酸序列的449位的色氨酸残基相当的氨基酸残基被苯丙氨酸残基所取代的变异。
[0067]
10、所述方法,其中,
[0068]
所述gh1-2蛋白为下述(a)、(b)或(c)所述的蛋白质:
[0069]
(a)包含seq id no.23所示的氨基酸序列的蛋白质;
[0070]
(b)包含在seq id no.23所示的氨基酸序列中包含1~10个氨基酸残基的取代、缺失、插入和/或添加的氨基酸序列,并且具有二糖水解活性的蛋白质;
[0071]
(c)包含相对于seq id no.23所示的氨基酸序列具有90%以上的同源性的氨基酸序列并且具有二糖水解活性的蛋白质。
[0072]
11、所述方法,其中,
[0073]
所述talaromyces cellulolyticus具有选自下述性质(a)~(d)中的任一种性质:
[0074]
(a)其经过了修饰,使其与非修饰菌株相比gh1-2蛋白以外的β-葡萄糖苷酶的活性降低;
[0075]
(b)其经过了修饰,使其与非修饰菌株相比crea蛋白的活性降低;
[0076]
(c)其经过了修饰,使其与非修饰菌株相比yscb蛋白的活性降低;
[0077]
(d)它们的组合。
[0078]
12、所述方法,其中,
[0079]
所述β-葡萄糖苷酶为bgl3a蛋白。
[0080]
13、所述方法,其中,
[0081]
所述talaromyces cellulolyticus为源自talaromyces cellulolyticus s6-25菌株(nite bp-01685)或y-94菌株(ferm bp-5826)的修饰菌株。
[0082]
14、所述方法,其中,
[0083]
通过所述培养而使靶蛋白积蓄在所述培养基中。
[0084]
15、所述方法,其中,
[0085]
靶蛋白在利用所述表达诱导物质对在talaromyces cellulolyticus中发挥功能的诱导性启动子进行控制之下进行表达。
[0086]
16、所述方法,其中,
[0087]
所述启动子为cbhi启动子或cbhii启动子。
[0088]
17、所述方法,其中,
[0089]
靶蛋白作为与在talaromyces cellulolyticus中发挥功能的信号肽形成的融合蛋白而进行表达。
[0090]
18、所述方法,其中,
[0091]
靶蛋白为纤维素酶。
[0092]
19、所述方法,其进一步包含下述工序:
[0093]
通过利用了二糖合成酶并且从糖原料出发的酶转化而制造所述龙胆二糖。
[0094]
20、所述方法,其中,
[0095]
所述酶转化通过使含有二糖合成酶的大肠杆菌的菌体与糖原料接触而实施。
[0096]
21、所述方法,其中,
[0097]
所述糖原料为选自葡萄糖、纤维二糖和纤维素中的1种或1种以上的糖原料。
[0098]
22、一种二糖的制造方法,其包含下述工序:
[0099]
通过使含有二糖合成酶的大肠杆菌(escherichia coli)的菌体与糖原料接触而
生成二糖。
[0100]
23、所述方法,其中,
[0101]
所述二糖合成酶为β-葡萄糖苷酶。
[0102]
24、所述方法,其中,
[0103]
所述β-葡萄糖苷酶为gh1-2蛋白或bgl3a蛋白。
[0104]
25、所述方法,其中,
[0105]
所述二糖合成酶为下述(a)、(b)或(c)所述的蛋白质:
[0106]
(a)包含seq id no.23或38所示的氨基酸序列的蛋白质;
[0107]
(b)包含在seq id no.23或38所示的氨基酸序列中包含1~10个氨基酸残基的取代、缺失、插入和/或添加的氨基酸序列,并且具有对由糖原料合成二糖的反应进行催化的活性的蛋白质;
[0108]
(c)包含相对于seq id no.23或38所示的氨基酸序列具有90%以上的同源性的氨基酸序列,并且具有对由糖原料合成二糖的反应进行催化的活性的蛋白质。
[0109]
26、所述方法,其中,
[0110]
所述二糖为选自龙胆二糖、纤维二糖、昆布二糖和槐糖中的1种或1种以上的二糖。
[0111]
27、所述方法,其中,
[0112]
所述二糖包含龙胆二糖。
[0113]
28、所述方法,其中,
[0114]
所述糖原料为选自葡萄糖、纤维二糖和纤维素中的1种或1种以上的糖原料。
[0115]
29、所述方法,其中,
[0116]
所述糖原料包含葡萄糖。
附图说明
[0117]
[图1]表示将基于t.cellulolyticus f09pyr+菌株和f09δgh1-2菌株的纤维素基质(solka-floc)作为碳源的纤维素酶生产的结果的图(照片)。
[0118]
[图2]表示将基于t.cellulolyticus f09pyr+菌株和f09δgh1-2菌株的纤维素基质(cmc)作为碳源的纤维素酶晕圈测定(cellulase halo assay)的结果的图(照片)。
[0119]
[图3]表示t.cellulolyticus的gh1-2在大肠杆菌(e.coli)中的异源表达的结果的图(照片)。
[0120]
[图4]表示基于t.cellulolyticus的gh1-2的从葡萄糖出发的β连接性的葡萄糖寡糖类的生产的结果的图。
[0121]
[图5]表示基于t.cellulolyticus f09pyr+菌株和f09δgh1-2菌株中的β连接性的葡萄糖寡糖的纤维素酶生产诱导的结果的图(照片)。
[0122]
[图6]表示基于t.cellulolyticus的gh1-2的从纤维二糖出发的β连接性的葡萄糖寡糖类的生产的结果的图(照片)。
[0123]
[图7]表示基于t.cellulolyticus的gh1-2的从纤维二糖出发的β连接性的葡萄糖寡糖类的生产的结果的图。
[0124]
[图8]表示基于表达t.cellulolyticus的gh1-2的大肠杆菌(e.coli)菌体的从葡萄糖出发的龙胆二糖的生产的结果的图。
cellulolyticus称为“本发明的微生物”。
[0145]
<1-1>表达诱导物质
[0146]
就表达诱导物质而言,只要是可以诱导本发明的微生物中靶蛋白的表达的物质,就没有特别限制。作为表达诱导物质,可举出包含葡萄糖作为构成糖的2残基或以上的长度的糖。典型性地,表达诱导物质可以仅包含葡萄糖作为构成糖。作为表达诱导物质,具体而言,可举出:葡萄糖的二糖(通过2分子的葡萄糖构成的二糖)、纤维寡糖、纤维素。作为二糖,可举出β连接性的二糖。作为β连接性的葡萄糖的二糖,具体而言,可举出:龙胆二糖、纤维二糖、昆布二糖、槐糖。作为纤维寡糖,例如可举出:纤维二糖、纤维三糖、纤维四糖。作为纤维素,例如可举出可以作为碳源使用的纤维素类基质。作为纤维素类基质,具体而言,例如可举出:微晶纤维素(avicel)、滤纸、废纸、纸浆、木材、稻草(rice straw)、麦秆(straw)、稻壳、米糠、麦麸、甘蔗渣、咖啡渣、茶渣。纤维素类基质在经过水热分解处理、酸处理、碱处理、蒸煮、爆碎、粉碎等的预处理之后可以用作表达诱导物质。作为市售的适宜的纤维素类基质,可举出solka floc(international fiber corp,north tonawanda,ny,u.s.a)。作为表达诱导物质,特别是可举出:龙胆二糖、纤维二糖、纤维素。作为表达诱导物质,可进一步特别举出龙胆二糖。作为表达诱导物质,可以使用1种物质,也可以组合使用2种或以上的物质。表达诱导物质可以根据表达中使用的启动子的种类、本发明的微生物所具有的修饰的种类等的各种条件进行适宜选择。在任何情况下,作为表达诱导物质,例如都可以选择龙胆二糖。此外,在本发明的微生物为修饰菌株的情况下,作为表达诱导物质,例如,可以选择通过组合本发明的微生物所具有的修饰与表达诱导物质的利用而增大靶蛋白的生产。
[0147]
就表达诱导物质而言,例如,其本身可以诱导靶蛋白的表达,也可以转化为别的物质而诱导靶蛋白的表达。具体而言,例如,表达诱导物质可以通过gh1-2蛋白的作用或通过gh1-2蛋白和其他酶的组合的作用,而将转化为别的物质并诱导靶蛋白的表达。作为其他酶,例如可举出纤维素酶。作为别的物质,例如可举出龙胆二糖、纤维二糖等的二糖。作为别的物质,可特别举出龙胆二糖。即,具体而言,例如,龙胆二糖以外的表达诱导物质可以通过gh1-2蛋白的作用或通过gh1-2蛋白和纤维素酶的组合的作用而转化为龙胆二糖并诱导靶蛋白的表达。
[0148]
<1-2>本发明的微生物
[0149]
本发明的微生物为具有靶蛋白生产能力的talaromyces cellulolyticus。需要说明的是,本发明的微生物的说明中,有时将本发明的微生物或用于构建其的talaromyces cellulolyticus称为“宿主”。
[0150]
<1-2-1>talaromyces cellulolyticus
[0151]
本发明的微生物为talaromyces cellulolyticus。talaromyces cellulolyticus的旧名为acremonium cellulolyticus。即,acremonium cellulolyticus通过系统分类的修订而被再分类为talaromyces cellulolyticus(fems microbiol.lett.,2014,351:32-41)。作为talaromyces cellulolyticus,具体而言,可举出:c1菌株(日本特开2003-135052)、cf-2612菌株(日本特开2008-271927)、tn菌株(ferm bp-685)、s6-25菌株(nite bp-01685)、y-94菌株(ferm bp-5826)和它们的衍生菌株。需要说明的是,“talaromyces cellulolyticus”总称为:在本申请的申请前、申请时和申请后的至少任一时间点被分类在talaromyces cellulolyticus中的真菌。即,例如,所述例举的菌株等的被分类在
talaromyces cellulolyticus中的菌株,即使在将来改变系统分类的情况下,也被视为属于talaromyces cellulolyticus。
[0152]
s6-25菌株原本于2013年8月8日被保藏在nite国际专利微生物保藏中心(日本千叶县木更津市上总镰足2-5-8 122号室,邮编292-0818),并于2013年11月15日基于布达佩斯条约而被移交至国际保藏中心,并被赋予保藏编号nite bp-01685。该菌株为由tn菌株(ferm bp-685)而得到的菌株,具有较高的纤维素酶生产能力。y-94菌株原本于1983年1月12日被保藏在通商产业省工业技术院生命工学工业技术研究所(现为nite国际专利微生物保藏中心,邮编292-0818,地址:日本千叶县木更津市上总镰足2-5-8 120号室),并于1997年2月19日基于布达佩斯条约而被移交至国际保藏中心,并被赋予保藏编号ferm bp-5826。
[0153]
例如,这些菌株可以从各菌株所保藏的保藏机关入手。
[0154]
作为本发明的微生物,可以将所述例举的菌株等的talaromyces cellulolyticus原样使用或进行适宜修饰后使用。即,例如,本发明的微生物可以为所述例举的菌株,也可以为源自所述例举的菌株的修饰菌株。具体而言,例如,本发明的微生物可以为源自s6-25菌株或y-94菌株的修饰菌株。
[0155]
<1-2-2>靶蛋白生产能力
[0156]
本发明的微生物具有靶蛋白生产能力。“具有靶蛋白生产能力的微生物”是指在表达诱导物质的存在下而具有生产靶蛋白的能力的微生物。“具有靶蛋白生产能力的微生物”具体而言可以指,在通过含有表达诱导物质的培养基而进行培养时,具有表达靶蛋白并在培养物中使靶蛋白积蓄至能够回收的程度的能力的微生物。具体而言,“在培养物中的积蓄”可以指在培养基中、菌体表层、菌体内或它们的组合中的积蓄。需要说明的是,将靶蛋白积蓄在菌体外(例如,培养基中、细胞表层)的情况称为靶蛋白的“分泌”或“分泌生产”。即,本发明的微生物可以具有靶蛋白的分泌生产能力(分泌生产靶蛋白的能力)。靶蛋白特别是可以积蓄在培养基中。就靶蛋白的积蓄量而言,例如,作为培养物中的积蓄量,可以为10μg/l以上、1mg/l以上、100mg/l以上或1g/l以上。本发明的微生物可以具有1种靶蛋白的生产能力,也可以具有2种或以上的靶蛋白的生产能力。
[0157]
本发明的微生物可以是本来就具有靶蛋白生产能力的物质,也可以是以具有靶蛋白生产能力的方式进行了修饰的物质。典型性地,本发明的微生物可以是本来就具有纤维素酶生产能力(在表达诱导物质的存在下生产纤维素酶的能力)的物质。此外,本发明的微生物可以是以使本来所具有的靶蛋白生产能力增强的方式进行了修饰的物质。就具有靶蛋白生产能力的微生物而言,例如,可以通过如上所述地赋予talaromyces cellulolyticus以靶蛋白生产能力,或,通过如上所述地增强talaromyces cellulolyticus的靶蛋白生产能力而取得。就靶蛋白生产能力而言,例如,可以通过靶蛋白的表达用的基因构建体的导入、其它提高靶蛋白生产能力的修饰的导入或它们的组合而进行赋予或增强。
[0158]
本发明的微生物依靠具有至少靶蛋白的表达用的基因构建体而具有靶蛋白生产能力。具体而言,本发明的微生物可以通过具有靶蛋白的表达用的基因构建体,或通过具有靶蛋白的表达用的基因构建体和其他性质的组合,而具有靶蛋白生产能力。即,本发明的微生物具有靶蛋白的表达用的基因构建体。本发明的微生物可以具有1拷贝的靶蛋白的表达用的基因构建体,也可以具有2拷贝或以上的靶蛋白的表达用的基因构建体。本发明的微生物可以具有1种靶蛋白的表达用的基因构建体,也可以具有2种或以上的靶蛋白的表达用的
基因构建体。靶蛋白的表达用的基因构建体的拷贝数和种类数可以分别称为靶蛋白基因的拷贝数和种类数。
[0159]
本发明的微生物中,靶蛋白的表达用的基因构建体可以存在于质粒这样的在染色体外进行自主复制的载体上,也可以整合在染色体上。即,就本发明的微生物而言,例如,可以在载体上具有靶蛋白的表达用的基因构建体,换而言之,可以具有包含靶蛋白的表达用的基因构建体的载体。此外,就本发明的微生物而言,例如,可以在染色体上具有靶蛋白的表达用的基因构建体。在本发明的微生物具有2个或以上的靶蛋白的表达用的基因构建体的情况下,这些基因构建体只要以能够制造靶蛋白的方式保持在本发明的微生物中即可。例如,这些基因构建体可以全部保持在单个的表达载体上,也可以全部保持在染色体上。此外,这些基因构建体可以分别保持在多个表达载体上,也可以分别保持在单个或多个表达载体上和染色体上。
[0160]
本发明的微生物可以是本来就具有靶蛋白的表达用的基因构建体的物质,也可以是以具有靶蛋白的表达用的基因构建体的方式进行了修饰的物质。典型性地,本发明的微生物可以是本来就具有纤维素酶的表达用的基因构建体的物质。此外,本发明的微生物可以是代替本来就具有的靶蛋白的表达用的基因构建体而导入了靶蛋白的表达用的基因构建体的物质、或进一步导入了靶蛋白的表达用的基因构建体的物质。具有靶蛋白的表达用的基因构建体的微生物,可以通过如上所述在talaromyces cellulolyticus中导入靶蛋白的表达用的基因构建体而取得。
[0161]“靶蛋白的表达用的基因构建体”是指,以在表达诱导物质的存在下能够表达靶蛋白的方式构成的基因表达系统。也将靶蛋白的表达用的基因构建体称为“靶蛋白的表达系统”、“靶蛋白的表达单元”或“靶蛋白的表达盒”。靶蛋白的表达用的基因构建体在5
’
至3
’
方向上包含基于表达诱导物质的诱导性启动子序列和编码靶蛋白的碱基序列。也将启动子序列简称为“启动子”。也将编码氨基酸序列的碱基序列称为“基因”。例如,也将编码靶蛋白编码的碱基序列称为“编码靶蛋白的基因”或“靶蛋白基因”。靶蛋白基因只要连接在启动子的下游以受到基于同启动子的控制并表达靶蛋白即可。此外,靶蛋白的表达用的基因构建体可以在适当的位置上具有用于表达靶蛋白的有效的控制序列(操纵子、终止子等)以使它们发挥功能。需要说明的是,本发明中,除非特别说明,否则“靶蛋白基因的表达”、“靶蛋白的表达”、“靶蛋白的生成”、“靶蛋白的生产”可以相互同义地使用。靶蛋白的表达用的基因构建体可以根据靶蛋白的种类等的各种条件而进行适宜设计。
[0162]
启动子只要是在talaromyces cellulolyticus中发挥功能的基于表达诱导物质的诱导性物质,就没有特别限制。启动子可以是基于1种表达诱导物质的诱导性物质,也可以是基于2种或以上的表达诱导物质的诱导性物质。作为启动子,使用至少基于所选择的表达诱导物质的诱导性物质。“在talaromyces cellulolyticus中发挥功能的启动子”是指在talaromyces cellulolyticus中具有启动子活性,即基因的转录活性的启动子。“基于表达诱导物质的诱导性启动子”是指使连接在下游的基因在表达诱导物质的存在下而被诱导表达的启动子。具体而言,“表达诱导物质的存在下”可以指在培养基中存在表达诱导物质。“基因在表达诱导物质的存在下而被诱导表达”是指表达诱导物质存在下的基因的表达水平高于不存在表达诱导物质下的基因的表达水平。具体而言,“基因在表达诱导物质的存在下而被诱导表达”是指,例如,表达诱导物质存在下中的基因的表达水平可以是不存在表达
诱导物质下的基因的表达水平的2倍以上、3倍以上或4倍以上。“基因在表达诱导物质的存在下而被诱导表达”还包含下述情况:在表达诱导物质不存在的情况下基因不表达而在表达诱导物质存在的情况下基因表达的情况。就来自启动子的基因表达而言,例如,可以通过表达诱导物质而直接性地诱导,也可以通过从表达诱导物质产生的别的物质而间接性地诱导。
[0163]
启动子可以是源自宿主的启动子,也可以是源自异源的启动子。启动子可以是靶蛋白基因的固有的启动子,也可以是其他基因的启动子。作为启动子,可举出通过表达诱导物质而诱导纤维素酶生产的微生物的纤维素酶基因的启动子。作为启动子,具体而言,可举出talaromyces cellulolyticus的纤维素酶基因的启动子。作为纤维素酶基因,可举出cbhi基因(也称为cbh1基因)、cbhii基因(也称为cbh2基因)。即,作为启动子,可举出cbhi基因的启动子、cbhii基因的启动子。也可以将cbhi基因的启动子称为“cbhi启动子”或“cbh1启动子”。也可以将cbhii基因的启动子称为“cbhii启动子”或“cbh2启动子”。这些启动子例如均可以作为基于选自所述例举的表达诱导物质中的1种或以上的物质的诱导性启动子而发挥功能。具体而言,这些启动子均可以作为至少基于龙胆二糖、纤维二糖和/或纤维素的诱导性启动子而发挥功能。更具体而言,这些启动子均可以作为至少基于龙胆二糖的诱导性启动子(龙胆二糖诱导性启动子)而发挥功能。将talaromyces cellulolyticus的cbhi启动子和cbhii启动子的碱基序列分别表示为seq id no.49和50。即,就启动子而言,例如,可以是具有所述例举的启动子的碱基序列(例如seq id no.49或50的碱基序列)的启动子。此外,启动子可以是所述例举的启动子(例如具有seq id no.49或50的碱基序列的启动子)的保守性变体。即,例如,所述例举的启动子可以原样使用或进行适宜修饰后使用。“cbhi启动子”和“cbhii启动子”这样的术语除了所述例举的cbhi启动子和cbhii启动子之外,还包含它们的保守性变体。对于启动子的保守性变体,可以适用涉及后述的gh1-2基因的保守性变体的记载。例如,启动子如果保持原本的功能,则可以是具有相对于seq id no.49或50的碱基序列具有80%以上,优选为90%以上,更优选为95%以上,进一步优选为97%以上,特别优选为99%以上的同源性的碱基序列的dna。需要说明的是,基于表达诱导物质的诱导性启动子的“原本的功能”是指,在表达诱导物质的存在下而诱导表达连接在下游的基因的功能。就基于表达诱导物质的诱导性启动子的功能而言,例如,可以通过下述方式进行确认:对向培养基供给表达诱导物质所带来的基因的诱导表达进行确认。就基因的诱导表达而言,例如,可以使用报告基因进行确认。
[0164]
靶蛋白没有特别限制。靶蛋白可以是源自宿主的蛋白质,也可以是源自异源的蛋白质(异源蛋白质)。本发明中,“异源蛋白质”(heterologous protein)是指,对于生产该蛋白质的talaromyces cellulolyticus而言是外来性(exogenous)的蛋白质。就靶蛋白而言,例如,可以是源自微生物的蛋白质,可以是源自植物的蛋白质,可以是源自动物的蛋白质,可以是源自病毒的蛋白质,可以是具有人工设计的氨基酸序列的蛋白质。特别是,靶蛋白可以是人源性蛋白质。靶蛋白可以是单体蛋白质,也可以是多聚体蛋白质。多聚体蛋白质是指,作为包含2或以上的亚基的多聚体而存在的蛋白质。多聚体中,各亚基可以通过二硫键等的共价键而进行连接,可以通过氢键、疏水性相互作用等的非共价键而进行连接,也可以通过它们的组合而进行连接。优选在多聚体中包含1个或以上的分子间二硫键。多聚体可以是包含单一的种类的亚基的同源多聚体,也可以是包含2或以上的种类的亚基的异源多聚
体。需要说明的是,“靶蛋白是异源蛋白质”是指,在靶蛋白是异源多聚体蛋白质的情况下,只要构成多聚体的亚基的中至少1个的亚基是异源蛋白质即可。即,可以所有亚基都源自异源,也可以仅一部分亚基源自异源。靶蛋白可以是分泌性蛋白,也可以是非分泌性蛋白。分泌性蛋白可以是天然分泌性的蛋白质,也可以是天然非分泌性的蛋白质,优选为天然分泌性的蛋白质。需要说明的是,“蛋白质”中还包含寡肽、多肽等的称为肽的物质。
[0165]
作为靶蛋白,例如可举出:酶、生物活性蛋白、受体蛋白、作为疫苗使用的抗原蛋白、其它任意的蛋白质。
[0166]
作为酶,例如可举出:纤维素酶、谷胺酰转氨酶、蛋白质谷氨酰胺酶(protein glutaminase)、异麦芽糖葡聚糖酶(isomaltodextranase)、蛋白酶、内肽酶、外肽酶、氨基肽酶、羧基肽酶、胶原酶和几丁质酶等。
[0167]
本发明中,“纤维素酶”是催化使纤维素中包含的糖苷键水解的反应的酶的总称。作为纤维素酶,可举出:内切型纤维素酶(内切葡聚糖酶;ec 3.2.1.4)、外切型纤维素酶(纤维二糖水解酶;ec 3.2.1.91)、纤维二糖酶(β-葡萄糖苷酶;ec 3.2.1.21)。此外,根据活性测定中使用的基质,纤维素酶称为微晶纤维酶(avicelase)、滤纸纤维素酶(fpase)、羧甲基纤维素酶(cmcase)等。作为纤维素酶,例如,可举出:里氏木霉(trichoderma reesei)、talaromyces cellulolyticus等的真菌、热纤梭菌(clostridium thermocellum)等的细菌的纤维素酶。
[0168]
作为谷胺酰转氨酶,例如,可举出:streptoverticillium mobaraense ifo 13819(wo01/23591)、streptoverticillium cinnamoneum ifo 12852、streptove rticillium griseocarneum ifo 12776、streptomyces lydicus(wo9606931)等的放线菌、oomycetes(wo9622366)等的丝状菌的分泌型的谷胺酰转氨酶。作为蛋白质谷氨酰胺酶,例如可举出chryseobacterium proteolyticum的蛋白质谷氨酰胺酶(wo2005/103278)。作为异麦芽糖葡聚糖酶,例如可举出arthrobacter globiformis的异麦芽糖葡聚糖酶(wo2005/103278)。
[0169]
作为生物活性蛋白,例如可举出:生长因子(growth factor),激素,细胞因子,抗体相关分子。
[0170]
作为生长因子(growth factor),具体而言,例如可举出:表皮生长因子(epidermal growth factor;egf)、胰岛素样生长因子-1(insulin-like growth factor-1;igf-1)、转化生长因子(transforming growth factor;tgf)、神经生长因子(nerve growth factor;ngf)、脑源性神经营养因子(brain-derived neurotrophic factor;bdnf)、血管内皮生长因子(vesicular endothelial growth factor;vegf)、粒细胞集落刺激因子(granulocyte-colony stimulating factor;g-csf)、粒细胞巨噬细胞集落刺激因子(granulocyte-macrophage-colony stimulating factor;gm-csf)、血小板衍生生长因子(platelet-derived growth factor;pdgf)、促红细胞生成素(erythropoietin;epo)、血小板生成素(thrombopoietin;tpo)、酸性成纤维细胞生长因子(acidic fibroblast growth factor;afgf或fgf1)、碱基性成纤维细胞生长因子(basic fibroblast growth factor;bfgf或fgf2)、角质细胞生长因子(keratinocyto growth factor;kgf-1或fgf7、kgf-2或fgf10)、肝细胞生长因子(hepatocyte growth factor;hgf)。
[0171]
作为激素,具体而言,例如可举出:胰岛素、胰高血糖素、生长抑素(somatostatin)、人类生长激素(human growth hormone;hgh)、甲状旁腺激素
(parathyroid hormone;pth)、降钙素(calcitonin)、艾塞那肽(exenatide)。
[0172]
作为细胞因子,具体而言,例如可举出:白细胞介素、干扰素、肿瘤坏死因子(tumor necrosis factor;tnf)。
[0173]
需要说明的是,生长因子(growth factor)、激素和细胞因子可以不用相互严格区别。例如,生物活性蛋白可以属于选自生长因子(growth factor)、激素和细胞因子中的任一组,也可以属于选自这些中的多个组。
[0174]
此外,生物活性蛋白可以是蛋白质整体,也可以是其一部分。作为蛋白质的一部分,例如可举出具有生理活性的部分。作为具有生理活性的部分,具体而言,例如可举出包含甲状旁腺激素(parathyroid hormone;pth)的成熟体的n末端34氨基酸残基的生理活性肽特立帕肽(teriparatide)。
[0175]“抗体相关分子”是指,包含由选自构成完整抗体的结构域的单个结构域或2或者2以上结构域的组合组成的分子种的蛋白质。作为构成完整抗体的结构域,可举出:作为重链的结构域的vh、ch1、ch2和ch3、以及作为轻链的结构域的vl和cl。抗体相关分子只要包含所述分子种即可,可以是单体蛋白质,也可以是多聚体蛋白质。需要说明的是,在抗体相关分子为多聚体蛋白质的情况下,可以是包含单一种类的亚基的同源多聚体,也可以是包含2或2个以上种类的亚基的异源多聚体。作为抗体相关分子,具体而言,例如可举出:完整抗体、fab、f(ab
’
)、f(ab
’
)2、fc、包含重链(h链)与轻链(l链)的二聚体、fc融合蛋白、重链(h链)、轻链(l链)、单链fv(scfv)、sc(fv)2、二硫键fv(sdfv)、双链抗体(diabody)、vhh片段(nanobody(注册商标))。作为抗体相关分子,更具体而言,例如可举出曲妥珠单抗、nivolumab。
[0176]
受体蛋白没有特别限制,例如,可以是对于生物活性蛋白、其它生理活性物质的受体蛋白。作为其它生理活性物质,例如可举出多巴胺等的神经递质。此外,受体蛋白也可以是对应的配体未知的孤儿受体。
[0177]
作为疫苗使用的抗原蛋白,只要能够引起免疫应答,就没有特别限制,根据假定的免疫应答的对象而适宜选择即可。
[0178]
此外,作为其它蛋白质,可举出:肝型脂肪酸结合蛋白liver-type fattyacid-binding protein(lfabp)、荧光蛋白、免疫球蛋白结合蛋白、白蛋白、细胞外蛋白。作为荧光蛋白,可举出绿色荧光蛋白green fluorescent protein(gfp)。作为免疫球蛋白结合蛋白,可举出protein a、protein g、protein l。作为白蛋白,可举出人血清白蛋白。
[0179]
作为细胞外蛋白,可举出:纤连蛋白、玻连蛋白、胶原蛋白、骨桥蛋白、层粘连蛋白、它们的部分序列。层粘连蛋白是具有包含α链、β链和γ链的异三聚体结构的蛋白质。作为层粘连蛋白,可举出哺乳动物的层粘连蛋白。作为哺乳动物,可举出:人、猴子、黑猩猩等的灵长类、小鼠、大鼠、仓鼠、豚鼠等的啮齿类、兔子、马、牛、绵羊、山羊、猪、狗、猫等的其它各种哺乳动物。作为哺乳动物,特别是可举出人。层粘连蛋白的亚基链(即,α链、β链和γ链),可举出:5种α链(α1~α5)、3种β链(β1~β3)、3种γ链(γ1~γ3)。层粘连蛋白通过这些亚基链的组合而构成各种的同工型。作为层粘连蛋白,具体而言,例如可举出:层粘连蛋白111、层粘连蛋白121、层粘连蛋白211、层粘连蛋白213、层粘连蛋白221、层粘连蛋白311、层粘连蛋白321、层粘连蛋白332、层粘连蛋白411、层粘连蛋白421、层粘连蛋白423、层粘连蛋白511、层粘连蛋白521、层粘连蛋白523。作为层粘连蛋白的部分序列,可举出作为层粘连蛋白的e8
片段的层粘连蛋白e8。具体而言,层粘连蛋白e8,是具有包含α链的e8片段(α链e8)、β链的e8片段(β链e8)和γ链的e8片段(γ链e8)的异三聚体结构的蛋白质。层粘连蛋白e8的亚基链(即,α链e8、β链e8和γ链e8)也总称为“e8亚基链”。作为e8亚基链,可举出所述例举的层粘连蛋白亚基链的e8片段。层粘连蛋白e8通过这些e8亚基链的组合而构成各种同工型。作为层粘连蛋白e8,具体而言,例如可举出:层粘连蛋白111e8、层粘连蛋白121e8、层粘连蛋白211e8、层粘连蛋白221e8、层粘连蛋白332e8、层粘连蛋白421e8、层粘连蛋白411e8、层粘连蛋白511e8、层粘连蛋白521e8。
[0180]
靶蛋白基因可以原样利用或适宜修饰后利用。靶蛋白基因,例如,为了得到期望的活性而可以进行修饰。对于靶蛋白基因和靶蛋白的变体,可以援用对后述的gh1-2基因和gh1-2蛋白的保守性变体的记载。例如,靶蛋白基因可以以使得在编码的靶蛋白的氨基酸序列中包含1个或几个氨基酸的取代、缺失、插入和/或添加的方式而进行修饰。需要说明的是,源自生物种的特定的蛋白质不限于在该生物种中发现的蛋白质本身,还包含具有在该生物种中发现的蛋白质的氨基酸序列的蛋白质和它们的变体。这些变体可以在该生物种中发现,也可以没有发现。即,例如,“人源蛋白质”是指,不限于在人体中发现的蛋白质本身,还包含具有在人体中发现的蛋白质的氨基酸序列的蛋白质和它们的变体。此外,靶蛋白基因可以将任意的密码子用与其等价的密码子取代。例如,就靶蛋白基因而言,可以根据使用的宿主的密码子使用频率而进行修饰,以使得具有最适的密码子。
[0181]
就靶蛋白而言,除了所述例举的靶蛋白的氨基酸序列之外,还可以包含其他氨基酸序列。即,靶蛋白可以是与其他氨基酸序列形成的融合蛋白。“其他氨基酸序列”只要能得到期望的性质的靶蛋白,就没有特别限制。“其他氨基酸序列”可以根据其利用目的等的各种条件而适宜选择。作为“其他氨基酸序列”,例如可举出:信号肽(也称为信号序列)、肽标签、蛋白酶的识别序列。“其他氨基酸序列”,例如,可以与靶蛋白的n末端或者c末端、或两者连接。作为“其他氨基酸序列”,可以使用1种氨基酸序列,也可以组合使用2种或2种以上的氨基酸序列。
[0182]
信号肽,例如,可以在靶蛋白的分泌生产中利用。信号肽可以与靶蛋白的n末端连接。即,在一个方式中,靶蛋白的表达用的基因构建体在从5
’
至3
’
方向上可以包含基于表达诱导物质的诱导性启动子序列、编码信号肽的碱基序列和编码靶蛋白的碱基序列。在这种情况下,可以将编码靶蛋白的核酸序列连接至编码信号肽的核酸序列的下游,以使得靶蛋白作为与同信号肽形成的融合蛋白而进行表达。需要说明的是,在这样的融合蛋白中,信号肽和靶蛋白可以相邻,也可以不相邻。即,“靶蛋白作为与信号肽形成的融合蛋白而进行表达”不限于靶蛋白与信号肽相邻并作为与同信号肽形成的融合蛋白而进行表达的情况,还包含靶蛋白借助其他氨基酸序列而作为与信号肽形成的融合蛋白而进行表达的情况。在利用信号肽而分泌生产靶蛋白的情况下,通常,在分泌时剪切信号肽,可以将没有信号肽的靶蛋白分泌在菌体外。即,“靶蛋白作为与信号肽形成的融合蛋白而进行表达”或“靶蛋白包含信号肽”是指,只要靶蛋白在表达时构成与信号肽形成的融合蛋白即可,不需要最终得到的靶蛋白构成与信号肽形成的融合蛋白。
[0183]
信号肽只要是在talaromyces cellulolyticus中发挥功能物质就没有特别限制。“在talaromyces cellulolyticus中发挥功能的信号肽”是指,当与靶蛋白的n末端连接时,在talaromyces cellulolyticus中带来靶蛋白的分泌的肽。
[0184]
信号肽可以是源自宿主的信号肽,也可以是源自异源的信号肽。信号肽可以是靶蛋白的固有的信号肽,也可以是其他蛋白质的信号肽。作为信号肽,可举出微生物的分泌性纤维素酶的信号肽。作为信号肽,具体而言,可举出talaromyces cellulolyticus的分泌性纤维素酶的信号肽。作为分泌性纤维素酶,可举出:cbhi基因编码的cbhi蛋白质(也称为cbh1蛋白质)、cbhii基因编码的cbhii蛋白质(也称为cbh2蛋白质)。即,作为信号肽,可举出:cbhi蛋白质的信号肽、cbhii蛋白质的信号肽。也将cbhi蛋白质的信号肽称为“cbhi信号肽”或“cbh1信号肽”。也将cbhii蛋白质的信号肽称为“cbhii信号肽”或“cbh2信号肽”。将talaromyces cellulolyticus的cbhi信号肽的氨基酸序列示于seq id no.51中。即,信号肽,例如,可以是具有所述例举的信号肽的氨基酸序列(例如seq id no.51的氨基酸序列)的信号肽。此外,信号肽可以是所述例举的信号肽(例如具有seq id no.51的氨基酸序列的信号肽)的保守性变体。即,例如,所述例举的信号肽可以原样使用或适宜修饰后使用。“cbhi信号肽”和“cbhii信号肽”这样的术语,除了所述例举的cbhi信号肽和cbhii信号肽之外,还包含它们的保守性变体。对于信号肽的保守性变体,可以援用涉及后述的gh1-2蛋白的保守性变体的记载。例如,信号肽如果保持原本的功能,则可以是具有在seq id no.51的氨基酸序列中,1或者几个位置的1个或几个氨基酸被取代、缺失、插入和/或添加的氨基酸序列的肽。需要说明的是,信号肽的变体中的所述“1个或几个”是指,具体而言,优选为1~7个,更优选为1~5个,近一些优选为1~3个,特别优选为1~2个。此外,例如,信号肽如果保持原本的功能,则可以是具有相对于seq id no.51的氨基酸序列,具有80%以上,优选为90%以上,更优选为95%以上,进一步优选为97%以上,特别优选为99%以上的同源性的氨基酸序列的肽。需要说明的是,对于信号肽的“原本的功能”,是可以在与靶蛋白的n末端连接时带来靶蛋白的分泌的功能。信号肽的功能,例如,可以通过对向蛋白质的n末端的连接引起的该蛋白质的分泌进行确认来确认。
[0185]
作为肽标签,具体而言,可举出:his标签、flag标签、gst标签、myc标签、mbp(maltose binding protein)、cbp(cellulose binding protein)、trx(thioredoxin)、gfp(green fluorescent protein)、hrp(horseradish peroxidase)、alp(alkaline phosphatase)、抗体的fc区域。肽标签,例如,可以在表达而得的靶蛋白的检测、纯化中利用。
[0186]
作为蛋白酶的识别序列,具体而言,可举出:hrv3c蛋白酶识别序列、factor xa蛋白酶识别序列、protev蛋白酶识别序列。蛋白酶的识别序列,例如,可以在表达而得的靶蛋白的剪切中利用。具体而言,例如,在使靶蛋白作为与肽标签形成的融合蛋白而进行表达的情况下,通过将蛋白酶的识别序列导入靶蛋白与肽标签的连接部,而能够利用蛋白酶从表达而得的靶蛋白剪切肽标签,得到没有肽标签的靶蛋白。
[0187]
最终得到的靶蛋白的n末端区域可以与天然的蛋白质相同,也可以与天然的蛋白质不相同。例如,与天然的蛋白质比较,最终得到的靶蛋白的n末端区域可以额外添加或缺失1个或几个氨基酸。需要说明的是,所述“1个或几个”是指,根据目的靶蛋白的全长、结构等而不同,具体而言,优选为1~20个,更优选为1~10个,进一步优选为1~5个,特别优选为1~3个。
[0188]
此外,靶蛋白可以作为添加有前体结构部的蛋白质(前体蛋白质)而进行表达。在靶蛋白作为前体蛋白质而进行表达的情况下,最终得到的靶蛋白可以是前体蛋白质,也可
以不是。即,前体蛋白质可以剪切前体结构部而成为成熟蛋白质。就剪切而言,例如,可以通过蛋白酶而进行。在使用蛋白酶的情况下,从最终得到的蛋白质的活性这样的观点出发,前体蛋白质通常优选在与天然的蛋白质大致相同位置剪切,更优选在与天然的蛋白质完全相同位置剪切而得到与天然的蛋白质相同的成熟蛋白质。因此,通常,最优选在产生天然产生的成熟蛋白质相同的蛋白质的位置剪切前体蛋白质的特异性蛋白酶。然而,如上所述,最终得到的靶蛋白的n末端区域可以与天然的蛋白质不相同。例如,根据要生产的靶蛋白的种类、使用目的等,n末端比天然的蛋白质长或短1~几个氨基酸的蛋白质可能具有更适当的活性。可以在本发明中使用的蛋白酶除了dispase(boehringer mannheim公司制)这样的可商购之外,还包含微生物的培养液,例如从放线菌的培养液等得到的蛋白酶。这样的蛋白酶可以在未纯化状态下使用,也可以根据需要纯化至适当的纯度后使用。
[0189]
靶蛋白基因,例如,可以通过克隆而取得。克隆,例如,可以利用包含靶蛋白基因的基因组dna、cdna等的核酸。此外,就靶蛋白基因而言,例如,也可以通过基于其碱基序列而进行全合成来取得(gene,60(1),115-127(1987))。取得的靶蛋白基因可以原样利用或适宜修饰后利用。即,通过对靶蛋白基因进行修饰,而能够取得其变体。基因的修饰可以通过公知的手法而进行。例如,可以通过位点特异性突变法而在dna的目的位点导入目的变异。作为位点特异性突变法,可举出:使用pcr的方法(higuchi,r.,61,in pcr technology,erlich,h.a.eds.,stockton press(1989);carter,p.,meth.in enzymol.,154,382(1987))、使用噬菌体的方法(kramer,w.and frits,h.j.,meth.in enzymol.,154,350(1987);kunkel,t.a.et al.,meth.in enzymol.,154,367(1987))。或者,可以全合成靶蛋白基因的变体。此外,可以对取得的靶蛋白基因适宜进行启动子序列的导入等的修饰,取得靶蛋白的表达用的基因构建体。需要说明的是,靶蛋白的表达用的基因构建体的其他构成要素(例如,启动子序列)、靶蛋白的表达用的基因构建体也可以通过与靶蛋白基因同样的方式取得。
[0190]
基因的修饰可以通过公知的手法而进行。例如,可以通过位点特异性突变法而在dna的目的位点导入目的变异。作为位点特异性突变法,可举出:使用pcr的方法(higuchi,r.,61,in pcr technology,erlich,h.a.eds.,stockton press(1989);carter,p.,meth.in enzymol.,154,382(1987))、使用噬菌体的方法(kramer,w.and frits,h.j.,meth.in enzymol.,154,350(1987);kunkel,t.a.et al.,meth.in enzymol.,154,367(1987))。
[0191]
将靶蛋白的表达用的基因构建体导入talaromyces cellulolyticus的手法没有特别限制。“靶蛋白的表达用的基因构建体的导入”是指只要将靶蛋白的表达用的基因构建体保持在宿主中即可,具体而言,是将靶蛋白基因以使其可以表达的方式导入宿主。“靶蛋白的表达用的基因构建体的导入”,只要没有特别说明,则不限于将预先构建的靶蛋白的表达用的基因构建体一并导入宿主的情况,还包含将靶蛋白的表达用的基因构建体的一部分导入宿主,并且在宿主内构建靶蛋白的表达用的基因构建体的情况。例如,可以通过将宿主本来就具有的靶蛋白基因的启动子用基于表达诱导物质的诱导性启动子取代,而在染色体上构建靶蛋白的表达用的基因构建体。此外,例如,可以通过将宿主本来就具有的靶蛋白基因导入基于表达诱导物质的诱导性启动子的下游,而在染色体上构建靶蛋白的表达用的基因构建体。
[0192]
就靶蛋白的表达用的基因构建体而言,例如,可以使用包含靶蛋白的表达用的基因构建体的载体而导入宿主。也将包含靶蛋白的表达用的基因构建体的载体称为“靶蛋白的表达载体”。就靶蛋白的表达载体而言,例如,可以通过将靶蛋白的表达用的基因构建体与载体进行连接而进行构建。此外,例如,在载体具备基于表达诱导物质的诱导性启动子的情况下,靶蛋白的表达载体可以通过将靶蛋白基因连接至该启动子的下游而进行构建。用靶蛋白的表达载体而转化宿主,从而得到导入了同载体的转化体,即,可以将靶蛋白的表达用的基因构建体导入宿主。就载体而言,只要可以在宿主的细胞内中自主复制就没有特别限制。载体可以是1拷贝载体,可以是低拷贝载体,也可以是多拷贝载体。载体可以具备用于选择转化体的标记基因。载体也可以具备用于表达靶蛋白基因的基于表达诱导物质的诱导性启动子、终止子。
[0193]
此外,靶蛋白的表达用的基因构建体可以导入宿主的染色体中。向染色体的基因的导入,可以利用同源重组而进行。具体而言,用包含靶蛋白的表达用的基因构建体的重组dna而转化宿主,与宿主的染色体上的目的位点发生同源重组,从而能够将靶蛋白的表达用的基因构建体导入宿主的染色体上。在同源重组中使用的重组dna的结构只要以期望的方式发生同源重组则没有特别限制。例如,可以是包含靶蛋白的表达用的基因构建体的线状dna,用在靶蛋白的表达用的基因构建体的两端分别具备染色体上的取代对象位点的上游和下游的序列的线状dna转化宿主,在取代对象位点的上游和下游分别发生同源重组,从而能够将取代对象位点用靶蛋白的表达用的基因构建体取代。同源重组中使用的重组dna可以具备用于选择转化体的标记基因。需要说明的是,靶蛋白基因、基于表达诱导物质的诱导性启动子等,向靶蛋白的表达用的基因构建体的一部分的染色体的导入也可以以与向靶蛋白的表达用的基因构建体整体的染色体的导入同样的方式进行。
[0194]
标记基因可以根据宿主的营养缺陷型等的性状而适宜选择。例如,在宿主通过pyrf基因或pyrg基因的变异而表现出尿嘧啶(uracil)缺陷型的情况下,将pyrf基因或pyrg基因作为标记基因而使用,从而能够将尿嘧啶(uracil)缺陷型的互补(即非尿嘧啶(uracil)缺陷型)作为指标,选拔导入了目的修饰的菌株。此外,作为标记基因,可以使用潮霉素抗性基因等的药剂抗性基因。
[0195]
就转化而言,例如,可以通过通常在霉菌、酵母等的真核微生物的转化中使用的手法而进行。作为这样的手法,可举出原生质体法。
[0196]
<1-2-3>其它性质
[0197]
本发明的微生物只要不损害靶蛋白生产能力,就可以具有期望的性质(例如修饰)。具有期望的性质的本发明的微生物,例如,可以通过对所述例举的菌株等的talaromyces cellulolyticus进行修饰而取得。作为修饰,可举出提高talaromyces cellulolyticus的靶蛋白生产能力的修饰。作为修饰,具体而言,可举出:降低gh1-2蛋白的活性的修饰、将变异导入gh1-2蛋白的修饰、降低β-葡萄糖苷酶的活性的修饰、降低crea蛋白的活性的修饰、降低yscb蛋白的活性的修饰。这些修饰可以单独利用或适宜组合利用。用于构建本发明的微生物的修饰的实施顺序没有特别限制。
[0198]
例如,可以以使得gh1-2蛋白的活性降低,并且/或gh1-2基因具有“特定的变异”的方式对本发明的微生物进行修饰。具体而言,可以以使得与非修饰菌株比较,gh1-2蛋白的活性降低的方式对本发明的微生物进行修饰。更具体而言,例如,可以以使得gh1-2基因的
表达降低的方式对本发明的微生物进行修饰,也可以以使得gh1-2基因被破坏的方式而进行修饰。通过以使得gh1-2蛋白的活性降低,并且/或gh1-2基因具有“特定的变异”的方式对talaromyces cellulolyticus进行修饰,而能够提高talaromyces cellulolyticus的靶蛋白生产能力,即,能够使表达诱导物质的存在下的talaromyces cellulolyticus带来的靶蛋白的生产增大。通过以使得gh1-2蛋白的活性降低,并且/或gh1-2基因具有“特定的变异”的方式对talaromyces cellulolyticus进行修饰,具体而言,而可以增强表达诱导物质引起的靶蛋白的表达诱导。“表达诱导物质引起的靶蛋白的表达诱导通过修饰而增强”是指,例如,修饰菌株中的表达诱导物质带来的靶蛋白的生产增大的程度(例如,数量、比例)比非修饰菌株中的表达诱导物质带来的靶蛋白的生产增大的程度(例如,数量、比例)高。
[0199]
以下,对gh1-2蛋白和编码其的gh1-2基因进行说明。需要说明的是,以下的说明除了本发明的微生物中活性降低的gh1-2蛋白之外,还兼具本发明的二糖的制造方法中利用的gh1-2蛋白的说明。本发明的微生物中活性降低的gh1-2蛋白是经过修饰的talaromyces cellulolyticus所具有的gh1-2蛋白。
[0200]
gh1-2蛋白是β-葡萄糖苷酶(beta-glucosidase)。具体而言,gh1-2蛋白可以是被分类为glucoside hydrolase family 1(gh1)的β-葡萄糖苷酶。此外,具体而言,gh1-2蛋白可以是细胞内定位型的β-葡萄糖苷酶。gh1-2蛋白具有对从糖原料合成(生成)二糖的反应进行催化的活性和/或对使二糖水解而生成葡萄糖的反应进行催化的活性。也将前者的活性称为“二糖合成活性(二糖生成活性)”或“糖转移活性”。也将后者的活性称为“二糖水解活性”。典型性地,gh1-2蛋白可以同时具有二糖合成活性和二糖水解活性两者。
[0201]
此处所说的“二糖”是指葡萄糖的二糖(由2分子的葡萄糖而构成的二糖)。作为二糖,可举出β连接性的二糖。作为二糖,具体而言,可举出:龙胆二糖、纤维二糖、昆布二糖、槐糖。作为二糖,特别是可举出龙胆二糖。
[0202]
此处所说的“糖原料”是指,包含葡萄糖作为构成糖的糖,还包含葡萄糖本身。典型性地,糖原料可以仅包含葡萄糖作为构成糖。作为糖原料,例如可举出:葡萄糖、纤维寡糖、纤维素。作为纤维寡糖,例如可举出:纤维二糖、纤维三糖、纤维四糖。作为纤维素,例如可举出所述的纤维素类基质。作为糖原料,可特别举出:葡萄糖、纤维二糖、纤维素。作为糖原料,可进一步特别举出葡萄糖。
[0203]
gh1-2蛋白可以具有对从糖原料合成1种二糖的反应进行催化的活性,也可以具有对从糖原料合成2种或2种以上的二糖的反应进行催化的活性。此外,gh1-2蛋白可以具有对从1种糖原料合成二糖的反应进行催化的活性,也可以具有对从2种或2种以上的糖原料合成二糖的反应进行催化的活性。例如,gh1-2蛋白可以至少具有对从糖原料合成龙胆二糖的反应进行催化的活性。此外,例如,gh1-2蛋白可以至少具有对从葡萄糖和/或纤维二糖合成二糖的反应进行催化的活性。gh1-2蛋白特别是可以至少具有对从葡萄糖合成二糖的反应进行催化的活性。更特别地,gh1-2蛋白可以至少具有对从葡萄糖和/或纤维二糖合成龙胆二糖的反应进行催化的活性。更特别地,gh1-2蛋白可以至少具有对从葡萄糖合成龙胆二糖的反应进行催化的活性。
[0204]
需要说明的是,gh1-2蛋白可以单独具有对从糖原料合成二糖的反应进行催化的活性,也可以在与其他酶组合使用时具有对从糖原料合成二糖的反应进行催化的活性。因此,gh1-2蛋白的活性中所说的“糖原料”不限于糖原料本身,例如,可以是糖原料通过其他
酶而转化得到的物质(例如糖原料的分解物)。即,在一个方式中,“二糖合成活性”可以是对从糖原料或其分解物合成二糖的反应进行催化的活性。作为其他酶,例如可举出纤维素酶。具体而言,例如,在使用纤维素作为糖原料,组合使用gh1-2蛋白和纤维素酶的情况下,可以通过纤维素酶而使糖原料分解,通过gh1-2蛋白而从糖原料的分解物生产二糖。
[0205]
gh1-2蛋白可以具有对水解1种二糖的反应进行催化的活性,也可以具有对水解2种或2种以上的二糖的反应进行催化的活性。例如,gh1-2蛋白可以至少具有对水解龙胆二糖和/或纤维二糖的反应进行催化的活性。gh1-2蛋白特别是可以至少具有对水解龙胆二糖的反应进行催化的活性。
[0206]
本发明的微生物的说明中的“gh1-2蛋白的活性降低”只要没有特别说明,则是指至少gh1-2蛋白的二糖水解活性降低。即,本发明的微生物中活性降低的gh1-2蛋白至少具有二糖水解活性。在gh1-2蛋白的二糖水解活性降低的情况下,gh1-2蛋白的二糖合成活性可以降低,也可以不降低。在gh1-2蛋白的二糖水解活性降低的情况下,典型性地,gh1-2蛋白的二糖合成活性也可能一并降低。即,“gh1-2蛋白的活性降低”可以是指,典型性地,gh1-2蛋白的二糖水解活性和二糖合成活性两者均降低。
[0207]
需要说明的是,通过以使得gh1-2蛋白的活性完全消失的方式对talaromyces cellulolyticus进行修饰,而能够特别是使在龙胆二糖的存在下的靶蛋白的生产增大。通过以使得gh1-2蛋白的活性完全消失的方式对talaromyces cellulolyticus进行修饰,而具体而言,可以使龙胆二糖的存在下的靶蛋白的表达诱导增强。通过以使得gh1-2蛋白的活性完全消失的方式对talaromyces cellulolyticus进行修饰,而更具体而言,可以使龙胆二糖的存在下的靶蛋白的表达诱导选择性地增强。因此,在以使得gh1-2蛋白的活性完全消失的方式对本发明的微生物进行修饰的情况下,作为表达诱导物质,例如,可以选择龙胆二糖。本发明的微生物的说明中的“gh1-2蛋白的活性完全消失”,只要没有特别说明,则是指至少gh1-2蛋白的二糖合成活性完全消失。在gh1-2蛋白的二糖合成活性完全消失的情况下,gh1-2蛋白的二糖水解活性可以完全消失,也可以不完全消失。在gh1-2蛋白的二糖合成活性完全消失的情况下,典型性地,gh1-2蛋白的二糖水解活性也可能一并完全消失。即,典型性地,“gh1-2蛋白的活性完全消失”可以指gh1-2蛋白的二糖合成活性和二糖水解活性两者均完全消失。
[0208]
作为gh1-2基因和gh1-2蛋白,可举出:talaromyces cellulolyticus等的真菌、其它微生物等的各种生物的物质。各种生物具有的gh1-2基因的碱基序列和由这些编码的gh1-2蛋白的氨基酸序列,例如,可以从ncbi(national center for biotechnology information)等的公开数据库取得。分别将talaromyces cellulolyticus y-94菌株的gh1-2基因(包含内含子)的碱基序列和该基因编码的gh1-2蛋白的氨基酸序列示于seq id no.1和23中。此外,将talaromyces cellulolyticus y-94菌株的gh1-2基因的cdna的碱基序列示于seq id no.22中。即,gh1-2基因,例如,可以是具有seq id no.1或22所示的碱基序列的基因。此外,gh1-2蛋白,例如,可以是具有seq id no.23所示的氨基酸序列的蛋白质。需要说明的是,“具有(氨基酸或碱基)序列”这样的表达包含该“包含(氨基酸或碱基)序列”的情况和该“由(氨基酸或碱基)序列组成”的情况。
[0209]
gh1-2基因只要保持原本的功能,则可以是所述例举的gh1-2基因(例如,具有seq id no.1或22所示的碱基序列的基因)的变体。同样,gh1-2蛋白只要保持原本的功能,则可
以是所述例举的gh1-2蛋白(例如,具有seq id no.23所示的氨基酸序列的蛋白质)的变体。有时也将这样的保持原本的功能的变体称为“保守性变体”。在本发明中,“gh1-2基因”这样的术语不限于所述例举的gh1-2基因,还包含其保守性变体。同样,“gh1-2蛋白”这样的术语不限于所述例举的gh1-2蛋白,还包含其保守性变体。作为保守性变体,例如可举出:所述例举的gh1-2基因、gh1-2蛋白的同系物、人为性的修饰体。
[0210]“保持原本的功能”是指,基因或蛋白质的变体具有与原本的基因或蛋白质的功能(活性、性质)对应的功能(活性、性质)。即,“保持原本的功能”是指,在gh1-2基因的情况下,基因的变体编码保持原本的功能的蛋白质。此外,“保持原本的功能”是指,在gh1-2蛋白的情况下,蛋白质的变体具有二糖合成活性和/或二糖水解活性。需要说明的是,作为通过某种gh1-2蛋白及其变体而合成或水解的二糖和基质使用的糖原料均可以彼此相同,也可以不同。
[0211]
二糖合成活性可以通过将酶与基质(葡萄糖等的糖原料)进行培养(incubate),测定酶和基质依赖性的产物(龙胆二糖等的二糖)的生成而进行测定。二糖水解活性可以通过将酶与基质(龙胆二糖等的二糖)进行培养(incubate),测定酶和基质依赖性的产物(葡萄糖)的生成而进行测定。产物的生成可以通过离子交换色谱法等的化合物的检测或鉴定中使用的公知的手法而进行确认。
[0212]
以下,将举例表示保守性变体。
[0213]
就gh1-2基因的同系物或gh1-2蛋白的同系物而言,例如,可以通过使用所述例举的gh1-2基因的碱基序列或所述例举的gh1-2蛋白的氨基酸序列作为查询序列的blast检索、fasta检索而从公开数据库容易地取得。此外,就gh1-2基因的同系物而言,例如,可以以talaromyces cellulolyticus等的生物的染色体为模板,通过使用基于这些公知的gh1-2基因的碱基序列而制备的寡核苷酸作为引物的pcr而取得。
[0214]
gh1-2基因只要保持原本的功能,则可以是编码具有在所述例举的gh1-2蛋白的氨基酸序列(例如,seq id no.23所示的氨基酸序列)中,1或者几个的位置上的1个或几个氨基酸被取代、缺失、插入和/或添加的氨基酸序列的蛋白质的基因。需要说明的是,所述“1个或几个”,根据氨基酸残基的蛋白质的立体结构中的位置、氨基酸残基的种类而不同,具体而言,例如为1~50个、1~40个、1~30个,优选为1~20个,更优选为1~10个,进一步优选为1~5个,特别优选为1~3个。
[0215]
所述的1或者几个的氨基酸的取代、缺失、插入和/或添加是正常保持蛋白质的功能的保守性变异。代表性的保守性变异是保守性取代。保守性取代是指,在取代位点为芳香族氨基酸的情况下,是在phe、trp、tyr间相互取代的变异,在取代位点为疏水性氨基酸的情况下,是在leu、ile、val间相互取代的变异,在为极性氨基酸的情况下,是在gln、asn间相互取代的变异,在为碱基性氨基酸的情况下,是在lys、arg、his间相互取代的变异,在为酸性氨基酸的情况下,是在asp、glu间相互取代的变异,在为具有羟基的氨基酸的情况下,是在ser、thr间相互取代的变异。作为被认为是保守性取代的取代,具体而言,可举出:从ala向ser或thr的取代,从arg向gln、his或lys的取代、从asn向glu、gln、lys、his或asp的取代、从asp向asn、glu或gln的取代、从cys向ser或ala的取代、从gln向asn、glu、lys、his、asp或arg的取代、从glu向gly、asn、gln、lys或asp的取代、从gly向pro的取代、从his向asn、lys、gln、arg或tyr的取代、从ile向leu、met、val或phe的取代、从leu向ile、met、val或phe的取代、从
lys向asn、glu、gln、his或arg的取代、从met向ile、leu、val或phe的取代、从phe向trp、tyr、met、ile或leu的取代、从ser向thr或ala的取代、从thr向ser或ala的取代、从trp向phe或tyr的取代、从tyr向his、phe或trp的取代和从val向met、ile或leu的取代。此外,所述的这样的氨基酸的取代、缺失、插入或添加,包含因基于源自基因的生物的个体差、物种的差异的情况等的天然产生的变异(mutant或variant)而产生的情况。
[0216]
此外,gh1-2基因只要保持原本的功能,则可以是编码具有下述氨基酸序列的蛋白质的基因,所述氨基酸序列对整个所述例举的gh1-2蛋白的氨基酸序列(例如,seq id no.23所示的氨基酸序列)具有80%以上,优选为90%以上,更优选为95%以上,进一步优选为97%以上,特别优选为99%以上的同源性的。需要说明的是,在本说明书中,“同源性”(homology)意味着“同一性”(identity)。
[0217]
此外,gh1-2基因只要保持原本的功能,则可以是在严格条件下与所述例举的gh1-2基因的碱基序列(例如,seq id no.1或22所示的碱基序列)的互补序列或可以从同互补序列制备的探针进行杂交的dna。“严格条件”是指,形成所谓的特异性的杂交,不形成非特异性的杂交的条件。如果显示一个实例,可举出:同源性较高的dna彼此杂交,例如,具有80%以上,优选为90%以上,更优选为95%以上,进一步优选为97%以上,特别优选为99%以上的同源性的dna彼此杂交,并且同源性较低的dna彼此不杂交的条件,或者与作为通常的southern杂交的洗涤的条件的60℃,1
×
ssc,0.1%sds,优选为60℃,0.1
×
ssc,0.1%sds,更优选为68℃,0.1
×
ssc,0.1%sds相当的盐浓度和温度下,进行1次,优选为2~3次清洗的条件。
[0218]
所述探针,例如,可以是基因的互补序列的一部分。这样的探针可以通过将基于公知的基因的碱基序列而制备的寡核苷酸作为引物,将包含这些碱基序列的dna片段设为模板的pcr而进行制备。作为探针,例如,可以使用300bp左右的长度的dna片段。在这样的情况下,作为杂交的洗涤的条件,可举出:50℃,2
×
ssc,0.1%sds。
[0219]
此外,gh1-2基因可以是将任意的密码子用与其等价的密码子取代而得到基因。即,gh1-2基因可以是基于密码子的简并的所述例举的gh1-2基因的变体。例如,就gh1-2基因而言,可以根据使用的宿主的密码子使用频率而进行修饰,以使得具有最适的密码子。
[0220]
2个序列间的序列同源性的百分比,例如,可以使用数学性算法来确定。作为这样的数学性算法的非限定的实例,可举出:myers and miller(1988)cabios 4:11-17的算法,smith et al(1981)adv.appl.math.2:482的局部相同算法、needleman and wunsch(1970)j.mol.biol.48:443-453的相同比对算法、检索pearson and lipman(1988)proc.natl.acad.sci.85:2444-2448的类似性的方法、karlin and altschul(1993)proc.natl.acad.sci.usa 90:5873-5877中记载的那样的改良的karlin and altschul(1990)proc.natl.acad.sci.usa 87:2264的算法。
[0221]
可以利用基于这些数学性算法的程序,进行用于确定序列同源性的序列比较(校准)。程序可以适宜通过计算机实行。作为这样的程序,没有特别限定,可举出:pc/gene程序的clustal(可从intelligenetics,mountain view,calif.入手)、align程序(version 2.0)以及wisconsin genetics software package,version 8(可从genetics computer group(gcg),575science drive,madison,wis.,usa入手)的gap、bestfit、blast、fasta和tfasta。使用了这些程序的校准,例如,可以使用初期参数而进行。对于clustal程序,良好
地记载在higgins et al.(1988)gene 73:237-244、higgins et al.(1989)cabios 5:151-153、corpet et al.(1988)nucleic acids res.16:10881-90、huang et al.(1992)cabios 8:155-65和pearson et al.(1994)meth.mol.biol.24:307-331中。
[0222]
为了得到与编码对象的蛋白质的核苷酸序列具有同源性的核苷酸序列,具体而言,例如,可以通过blastn程序,在分数=100,字长=12下进行blast核苷酸检索。为了得到与对象的蛋白质具有同源性的氨基酸序列,具体而言,例如,可以通过blastx程序,在分数=50,字长=3下进行blast蛋白质检索。对于blast核苷酸检索、blast蛋白质检索,参照http://www.ncbi.nlm.nih.gov。此外,为了得到为了比较而添加了间隙的校准,可以利用gapped blast(blast 2.0)。此外,可以利用psi-blast(blast 2.0)检测序列间的离间关系并进行反复检索。对于gapped blast和psi-blast,参照altschul et al.(1997)nucleic acids res.25:3389。在利用blast、gapped blast或psi-blast的情况下,例如,可以使用各程序(例如,针对核苷酸序列的blastn,针对氨基酸序列的blastx)的初期参数。校准可以手动执行。
[0223]
2个序列间的序列同源性作为当将2个序列以变成最大一致的方式进行排列时2个序列间一致的残基的比例而算出。需要说明的是,氨基酸序列间的“同源性”,具体而言,只要没有特别说明,则是指使用通过blastp而默认设定的scoring parameters(matrix:blosum62;gap costs:existence=11,extension=1;compositional adjustments:conditional compositional score matrix adjustment)算出的氨基酸序列间的同源性。此外,碱基序列间的“同源性”,具体而言,只要没有特别说明,则是指使用通过blastn而默认设定的scoring parameters(match/mismatch scores=1,-2;gap costs=linear)而算出的碱基序列间的同源性。
[0224]
需要说明的是,与所述的基因、蛋白质的变体有关的记载也可以援用在靶蛋白等的任意的蛋白质和编码它们的基因中。
[0225]
gh1-2蛋白的活性可以通过下述方式降低,例如,通过弱化gh1-2基因的表达,或通过破坏gh1-2基因。此外,在一个方式中,例如,可以以具有“特定的变异”的方式修饰gh1-2基因而降低gh1-2蛋白的活性。这样的降低gh1-2蛋白的活性的手法可以单独使用或适宜组合使用。
[0226]
本发明的微生物可以以使得gh1-2基因具有“特定的变异”的方式进行修饰。
[0227]“特定的变异”是使talaromyces cellulolyticus的靶蛋白生产能力提高的变异。“特定的变异”是指,在gh1-2基因的情况下,gh1-2基因的碱基序列中的变化。通过“特定的变异”,而可以发生编码的gh1-2蛋白的氨基酸序列中变化。因此,“特定的变异”也可以是指,对于gh1-2蛋白,通过gh1-2基因中的“特定的变异”而发生的gh1-2蛋白的氨基酸序列中的变化。即,“gh1-2基因具有“特定的变异
””
可以解读为该基因编码的gh1-2蛋白具有“特定的变异”。
[0228]
也将具有“特定的变异”的gh1-2蛋白称为“变异型gh1-2蛋白”。此外,也将编码变异型gh1-2蛋白的基因,即具有“特定的变异”的gh1-2基因称为“变异型gh1-2基因”。
[0229]
也将不具有“特定的变异”的gh1-2蛋白称为“野生型gh1-2蛋白”。此外,也将编码野生型gh1-2蛋白的基因,即不具有“特定的变异”的gh1-2基因称为“野生型gh1-2基因”。作为野生型gh1-2基因或野生型gh1-2蛋白,例如可举出:所述例举的gh1-2基因或gh1-2蛋白
et al.,journal of molecular biology,198(2),327-37.1987)。
[0238]
就变异型gh1-2基因而言,例如,可以通过以使得编码的gh1-2蛋白具有“特定的变异”的方式对野生型gh1-2基因进行修饰而取得。就成为修饰的源头的野生型gh1-2基因而言,例如,可以通过从具有野生型gh1-2基因的生物的克隆或通过化学合成而取得。或,变异型gh1-2基因可以不借助野生型gh1-2基因而取得。就变异型gh1-2基因而言,例如,可以通过从具有变异型gh1-2基因的生物的克隆或通过化学合成而直接取得。取得的变异型gh1-2基因可以原样利用或进一步修饰后利用。基因的修饰可以通过公知的手法而进行。例如,可以通过位点特异性突变法,而在dna的目的位点导入目的变异。成为修饰的源头的野生型gh1-2基因或变异型gh1-2基因可以是源自宿主的,也可以不是。
[0239]
以使得gh1-2基因具有“特定的变异”的方式对talaromyces cellulolyticus进行修饰的手法没有特别限制。“以使得gh1-2基因具有“特定的变异”的方式对talaromyces cellulolyticus进行修饰”,具体而言,可以是以使得代替天生(native)的野生型gh1-2基因而具有变异型gh1-2基因的方式对talaromyces cellulolyticus进行修饰。“talaromyces cellulolyticus代替天生的野生型gh1-2基因而具有变异型gh1-2基因”可以是,talaromyces cellulolyticus具有变异型gh1-2基因,不再具有正常发挥功能的天生的野生型gh1-2基因(即,天生的野生型gh1-2基因经过修饰,而使得无法正常发挥功能)。“天生的野生型gh1-2基因”可以是指,本来存在于talaromyces cellulolyticus的野生型gh1-2基因。例如,通过将变异型gh1-2基因导入talaromyces cellulolyticus中,而能够以使得gh1-2基因具有“特定的变异”的方式对talaromyces cellulolyticus进行修饰。在这种情况下,对存在于talaromyces cellulolyticus的染色体等中的天生的野生型gh1-2基因进行修饰(例如,破坏、损失),以使得通过与变异型gh1-2基因的导入的组合而使talaromyces cellulolyticus的靶蛋白生产能力提高。例如,天生的野生型gh1-2基因可以被变异型gh1-2基因取代,也可以独立于变异型gh1-2基因的导入而进行破坏或损失。或者,例如,通过将“特定的变异”导入存在于talaromyces cellulolyticus的染色体等中的野生型gh1-2基因(例如,天生的野生型gh1-2基因),而可以以使得gh1-2基因具有“特定的变异”方式对talaromyces cellulolyticus进行修饰。就变异而言,例如,可以通过自然变异、变异处理或基因工程而导入染色体等中存在的基因。
[0240]
此外,例如,可以对本发明的微生物进行修饰,使得β-葡萄糖苷酶的活性降低。具体而言,可以对本发明的微生物进行修饰,使其与非修饰菌株相比β-葡萄糖苷酶的活性降低。更具体而言,例如,可以对本发明的微生物进行修饰,使得β-葡萄糖苷酶基因的表达降低,也可以对本发明的微生物进行修饰,使得β-葡萄糖苷酶基因被破坏。“β-葡萄糖苷酶”是指,具有二糖合成活性和/或二糖水解活性的蛋白质(酶)。典型性地,β-葡萄糖苷酶可以同时具有二糖合成活性和二糖水解活性两者。对于β-葡萄糖苷酶的活性,可以援用对gh1-2蛋白的活性的记载。例如,本发明的微生物的说明中的“β-葡萄糖苷酶的活性降低”,只要没有特别说明,则是指至少β-葡萄糖苷酶的二糖水解活性降低。即,本发明的微生物中活性降低的β-葡萄糖苷酶至少具有二糖水解活性。对talaromyces cellulolyticus进行修饰,使得β-葡萄糖苷酶的活性降低,从而能够提高talaromyces cellulolyticus的靶蛋白生产能力,即,期待可以使表达诱导物质的存在下的talaromyces cellulolyticus带来的靶蛋白的生产增大。
[0241]
β-葡萄糖苷酶可以是细胞内定位型,也可以是细胞外分泌型。作为β-葡萄糖苷酶,可举出gh1-2蛋白以外的β-葡萄糖苷酶。作为gh1-2蛋白以外的β-葡萄糖苷酶,可举出bgl3a蛋白。具体而言,bgl3a可以是被分类为glucoside hydrolase family 3(gh3)的β-葡萄糖苷酶。此外,具体而言,bgl3a蛋白可以是细胞外分泌型的β-葡萄糖苷酶。
[0242]
作为β-葡萄糖苷酶基因和β-葡萄糖苷酶,可举出talaromyces cellulolyticus等的真菌、其它微生物等的各种生物的。各种生物具有的β-葡萄糖苷酶基因的碱基序列和由这些编码的β-葡萄糖苷酶的氨基酸序列,例如,可以从ncbi(national center for biotechnology information)等的公开数据库取得。分别将talaromyces cellulolyticus的bgl3a基因的cdna的碱基序列和该基因编码的bgl3a蛋白的氨基酸序列示于seq id no.48和38中。即,β-葡萄糖苷酶基因,例如,可以是具有seq id no.48所示的碱基序列的基因。此外,β-葡萄糖苷酶,例如,可以是具有seq id no.38所示的氨基酸序列的蛋白质。β-葡萄糖苷酶基因和β-葡萄糖苷酶可以分别是所述例举的β-葡萄糖苷酶基因和β-葡萄糖苷酶的保守性变体(例如,所述例举的bgl3a基因和bgl3a蛋白的保守性变体)。对于β-葡萄糖苷酶基因和β-葡萄糖苷酶的保守性变体,可以援用对gh1-2基因和gh1-2蛋白的保守性变体的记载。需要说明的是,“保持原本的功能”是指,在β-葡萄糖苷酶的情况下,蛋白质的变体具有二糖合成活性和/或二糖水解活性。
[0243]
需要说明的是,与所述β-葡萄糖苷酶有关的说明除了在本发明的微生物中活性降低的β-葡萄糖苷酶之外,还兼具本发明的二糖的制造方法中利用的β-葡萄糖苷酶的说明。在本发明的微生物中活性降低的β-葡萄糖苷酶是经过修饰的talaromyces cellulolyticus具有的β-葡萄糖苷酶。
[0244]
此外,例如,可以对本发明的微生物进行修饰,使得crea蛋白的活性降低。具体而言,可以对本发明的微生物进行修饰,使其与非修饰菌株比较crea蛋白的活性降低。更具体而言,例如,可以对本发明的微生物进行修饰,使得crea基因的表达降低,或使得crea基因被破坏。crea基因是编码参与分解代谢物阻遏的转录因子的基因。已知crea基因在丝状菌中参与纤维素酶的表达(mol gen genet.1996jun 24;251(4):451-60,biosci biotechnol biochem.1998dec;62(12):2364-70)。对talaromyces cellulolyticus进行修饰,使得crea蛋白的活性降低,从而能够提高talaromyces cellulolyticus的靶蛋白生产能力,即,期待可以使表达诱导物质的存在下的talaromyces cellulolyticus带来的靶蛋白的生产增大。
[0245]
将talaromyces cellulolyticus s6-25菌株的crea基因的碱基序列示于seq id no.47中。即,例如,crea基因可以是具有seq id no.47所示的碱基序列的基因。此外,例如,crea蛋白可以是具有通过seq id no.47所示的碱基序列而编码的氨基酸序列的蛋白质。crea基因和crea蛋白可以分别是所述例举的crea基因和crea蛋白的保守性变体。对于crea基因和crea蛋白的保守性变体可以援用对gh1-2基因和gh1-2蛋白的保守性变体的记载。需要说明的是,“保持原本的功能”是指,在crea蛋白的情况下,蛋白质的变体具有作为参与分解代谢物阻遏的转录因子的功能。
[0246]
此外,例如,可以对本发明的微生物进行修饰,使得yscb蛋白的活性降低。具体而言,可以对本发明的微生物进行修饰,使其与非修饰菌株相比yscb蛋白的活性降低。更具体而言,例如,可以对本发明的微生物进行修饰,使得yscb基因的表达降低,或使得yscb基因被破坏。yscb蛋白是蛋白酶。“蛋白酶”是指,具有对使蛋白质水解的反应进行催化的活性的
蛋白质。此外,也将该活性称为“蛋白酶活性”。对talaromyces cellulolyticus进行修饰,使得yscb蛋白的活性降低,从而能够提高talaromyces cellulolyticus的靶蛋白生产能力,即,期待可以使表达诱导物质的存在下的talaromyces cellulolyticus带来的靶蛋白的生产增大。
[0247]
分别将talaromyces cellulolyticus s6-25菌株的yscb基因(包含内含子)的碱基序列和该基因编码的yscb蛋白的氨基酸序列示于seq id no.62和69中。即,例如,yscb基因可以是具有seq id no.62所示的碱基序列的基因。此外,例如,yscb蛋白可以是具有seq id no.69所示的氨基酸序列的蛋白质。yscb基因和yscb蛋白可以分别是所述例举的yscb基因和yscb蛋白的保守性变体。对于yscb基因和yscb蛋白的保守性变体,可以援用对gh1-2基因和gh1-2蛋白的保守性变体的记载。需要说明的是,“保持原本的功能”是指,在yscb蛋白的情况下,蛋白质的变体具有蛋白酶活性。
[0248]
蛋白酶活性可以通过将酶与基质(蛋白质)进行培养(incubate),测定酶依赖性的基质的分解而进行测定。此外,蛋白酶活性可以使用市售的蛋白酶活性测定试剂盒来测定。
[0249]
<1-2-4>使蛋白质的活性降低的手法
[0250]
以下,对使gh1-2蛋白、bgl3a蛋白等的β-葡萄糖苷酶、crea蛋白、yscb蛋白等的蛋白质的活性降低的手法进行说明。
[0251]“蛋白质的活性降低”是指,与非修饰菌株相比该蛋白质的活性降低。具体而言,“蛋白质的活性降低”是指,与非修饰菌株相比该蛋白质在每个细胞中的活性降低。此处所说的“非修饰菌株”是指,未以使得靶蛋白的活性降低的方式而修饰的对照菌株。作为非修饰菌株,可举出野生菌株、亲本菌株。作为非修饰菌株,具体而言,可举出talaromyces cellulolyticus的说明中例举的菌株。即,在一个方式中,与talaromyces cellulolyticuss6-25菌株相比蛋白质的活性可以降低。此外,在别的方式中,与talaromyces cellulolyticusy-94菌株相比蛋白质的活性可以降低。需要说明的是,“蛋白质的活性降低”也包含该蛋白质的活性完全消失的情况。更具体而言,“蛋白质的活性降低”可以是指,与非修饰菌株相比,该蛋白质在每个细胞中的分子数降低,和/或,该蛋白质的每个分子的功能降低。即,“蛋白质的活性降低”这样的情况的“活性”,不限于蛋白质的催化剂活性,可以是指编码蛋白质的基因的转录量(mrna量)或翻译量(蛋白质的量)。需要说明的是,“蛋白质在每个细胞中的分子数降低”也包含该蛋白质完全不存在的情况。此外,“蛋白质的每个分子的功能降低”也包含该蛋白质的每个分子的功能完全消失的情况。蛋白质的活性的降低的程度,如果与非修饰菌株相比蛋白质的活性降低,则没有特别限制。蛋白质的活性,例如,可以降低至非修饰菌株的50%以下、20%以下、10%以下、5%以下或0%。
[0252]
这样的使蛋白质的活性降低的修饰,例如,可以通过使编码该蛋白质的基因的表达降低而达成。“基因的表达降低”是指,与非修饰菌株相比该基因的表达降低。“基因的表达降低”是指,具体而言,与非修饰菌株相比该基因在每个细胞中的表达量降低。“基因的表达降低”可以是指,更具体而言,基因的转录量(mrna量)降低,和/或,基因的翻译量(蛋白质的量)降低。“基因的表达降低”包含该基因完全不表达的情况。需要说明的是,也将“基因的表达降低”称为“基因的表达弱化”。基因的表达,例如,可以降低至非修饰菌株的50%以下、20%以下、10%以下、5%以下或0%。
[0253]
基因的表达的降低,例如,可以基于转录效率的降低,可以基于翻译效率的降低,
也可以基于它们的组合。基因的表达的降低,例如,可以通过对基因的表达调控序列进行修饰而达成。“表达调控序列”是影响启动子等的基因的表达的位点的总称。表达调控序列,例如,可以使用启动子检索载体、genetyx等的基因分析软件而进行确定。在对表达调控序列进行修饰的情况下,表达调控序列优选1碱基以上,更优选2碱基以上,特别优选3碱基以上被修饰。基因的转录效率的降低,例如,可以通过用更弱的启动子取代染色体上的基因的启动子而达成。“更弱的启动子”是指,基因的转录比原本存在的野生型的启动子弱的启动子。作为更弱的启动子,例如可举出诱导型的启动子。即,诱导型的启动子可以在非诱导条件下(例如,不存在诱导物质下)作为更弱的启动子而发挥功能。此外,可以使表达调控序列的一部分或全部的区域缺失(损失)。此外,基因的表达的降低,例如,可以通过操纵与表达控制有关的因子而达成。作为与表达控制有关的因子,可举出:与转录、翻译控制有关的低分子(诱导物质、阻碍物质等)、蛋白质(转录因子等)、核酸(sirna等)等。此外,基因的表达的降低,例如,也可以通过在基因的编码区域中导入使基因的表达降低这样的变异而达成。例如,将基因的编码区域的密码子替换为在宿主中以更低频率利用的同义密码子,因此能够使基因的表达降低。此外,例如,通过后述这样的基因的破坏而可以使基因的表达本身降低。
[0254]
此外,这样的使蛋白质的活性降低的修饰,例如,可以通过对编码该蛋白质的基因进行破坏而达成。“使基因破坏”是指,对该基因进行修饰,使得不产生正常发挥功能的蛋白质。“不产生正常发挥功能的蛋白质”包含从该基因完全不产生蛋白质的情况、从该基因产生每个分子的功能(活性、性质)降低或消失的蛋白质的情况。
[0255]
基因的破坏,例如,可以通过使染色体上的基因缺失(损失)而达成。“基因的缺失”是指,基因的编码区域的一部分或全部的区域的缺失。进一步,也可以使包含染色体上的基因的编码区域的前后的序列的整个基因缺失。基因的编码区域的前后的序列,例如,可以包含基因的表达调控序列。只要能够达成蛋白质的活性的降低,则缺失的区域可以是n末端区域(编码蛋白质的n末端侧的区域)、内部区域、c末端区域(编码蛋白质的c末端侧的区域)等的任一区域。通常,缺失的区域长的情况能够确实地使基因不活化。缺失的区域,例如,可以是基因的编码区域全长的10%以上、20%以上、30%以上、40%以上、50%以上、60%以上、70%以上、80%以上、90%以上或95%以上的长度的区域。此外,优选缺失的区域的前后的序列的阅读框不一致。可以通过阅读框的不一致而在缺失的区域的下游发生移码。在crea基因的情况下,具体而言,例如,通过使与seq id no.47的3262~4509位相当的部分缺失,而能够破坏该基因。
[0256]
此外,基因的破坏,例如,可以通过在染色体上的基因的编码区域中导入氨基酸取代(错义突变)、导入终止密码子(无义突变)或导入1~2碱基的添加或缺失(移码变异)等而达成(journal of biological chemistry 272:8611-8617(1997),proceedings of the national academy of sciences,usa 95 5511-5515(1998),journal of biological chemistry 26 116,20833-20839(1991))。
[0257]
此外,基因的破坏,例如,可以通过在染色体上的基因的编码区域中插入其他碱基序列而达成。插入位点可以是基因的任一区域,插入的碱基序列长的情况能够确实得使基因不活化。此外,优选插入位点的前后的序列的阅读框不一致。通过阅读框的不一致而可以在插入位点的下游发生移码。作为其他碱基序列,如果使编码的蛋白质的活性降低或消失
则没有特别限制,例如,可举出在标记基因、靶蛋白生产中有用的基因。
[0258]
特别是,可以实施基因的破坏,使得编码的蛋白质的氨基酸序列缺失(损失)。换而言之,这样的使蛋白质的活性降低的修饰,例如,可以通过使蛋白质的氨基酸序列缺失而达成,具体而言,可以通过以使其编码氨基酸序列缺失的蛋白质的方式对基因进行修饰而达成。需要说明的是,“蛋白质的氨基酸序列的缺失”是指,蛋白质的氨基酸序列的一部分或全部的区域的缺失。此外,“蛋白质的氨基酸序列的缺失”是指在蛋白质中原本的氨基酸序列变为不存在,也包含原本的氨基酸序列变为别的氨基酸序列的情况。即,例如,通过移码而变为别的氨基酸序列的区域可以被认为是缺失的区域。通过蛋白质的氨基酸序列的缺失,典型性地使蛋白质的全长短缩,但也可能存在蛋白质的全长不变化,或延长的情况。例如,通过使基因的编码区域的一部分或全部的区域的缺失,而能够在编码的蛋白质的氨基酸序列中,使该缺失的区域所编码的区域缺失。此外,例如,通过向基因的编码区域的终止密码子的导入,而能够在编码的蛋白质的氨基酸序列中,使在该导入位点的下游的区域所编码的区域缺失。此外,例如,通过基因的编码区域中的移码,而能够使该移码位点所编码的区域缺失。对于氨基酸序列的缺失中的缺失的区域的位置和长度,可以援用基因的缺失中的缺失的区域的位置和长度的说明。
[0259]
在对染色体上的基因进行如上所述的修饰的情况下,例如可以通过下述方式达成:制备经过修饰的破坏型基因,使其不产生正常发挥功能的蛋白质,用包含该破坏型基因的重组dna转化宿主,通过破坏型基因和染色体上的野生型基因而发生同源重组,从而用破坏型基因取代染色体上的野生型基因。此时,重组dna如果根据宿主的营养缺陷型等的性状而包含标记基因,则容易操纵。作为破坏型基因,可举出:缺失了基因的编码区域的一部分或全部的区域的基因、导入了错义突变的基因、导入了无义突变的基因、导入了移码变异的基因、导入了转座子、标记基因等的插入序列的基因。由破坏型基因而编码的蛋白质,即使生成,也具有与野生型蛋白质不同的立体结构,功能降低或消失。
[0260]
同源重组中使用的重组dna的结构,只要以期望的方式发生同源重组,则没有特别限制。例如为包含任意的序列的线状dna,用在该任意的序列的两端分别具备染色体上的取代对象位点的上游和下游的序列的线状dna转化宿主,分别使取代对象位点的上游和下游发生同源重组,从而能够在1步骤中用该任意的序列取代要取代对象位点。作为该任意的序列,例如可以使用包含标记基因的序列。
[0261]
标记基因可以根据宿主的营养缺陷型等的性状而适宜选择。例如,在宿主通过pyrf基因或pyrg基因的变异而表现出尿嘧啶(uracil)缺陷型的情况下,通过将pyrf基因或pyrg基因作为标记基因使用,而能够将尿嘧啶(uracil)缺陷型的互补(即非尿嘧啶(uracil)缺陷型)作为指标,选拔导入有目的修饰的菌株。此外,例如,在通过sc基因(sulfate permiase基因)的变异而表现出蛋氨酸缺陷型的情况下,通过将sc基因作为标记基因使用,而能够将蛋氨酸缺陷型的互补(即蛋氨酸非缺陷型)作为指标,选拔导入有目的修饰的菌株。此外,作为标记基因,可以使用潮霉素抗性基因等的药剂抗性基因。
[0262]
此外,这样的使蛋白质的活性降低的修饰,例如,可以通过突变处理而进行。作为突变处理,可举出:x射线的照射、紫外线的照射、以及基于n-甲基-n'-硝基-n-亚硝基胍(mnng)、甲磺酸乙酯(ems)和甲基磺酸甲酯(mms)等的变异剂的处理。
[0263]
可以通过对该蛋白质的活性进行测定来确认蛋白质的活性降低。gh1-2蛋白、
bgl3a蛋白等的β-葡萄糖苷酶、和yscb蛋白的活性例如可以通过如上所述的方式进行测定。例如,crea蛋白的活性可以通过对分解代谢物阻遏的程度进行测定而测定。例如,分解代谢物阻遏的程度可以通过对包含葡萄糖作为碳源的培养条件下的纤维素酶生产进行测定而测定。即,具体而言,例如,可以通过将包含葡萄糖作为碳源的培养条件下的纤维素酶生产的提高作为指标来确认crea蛋白的活性降低。
[0264]
也可以通过对编码该蛋白质的基因的表达降低进行确认来确认蛋白质的活性降低。基因的表达降低也可以通过下述方式确定:通过对该基因的转录量降低进行确认或通过对从该基因表达的蛋白质的量降低进行确认。
[0265]
基因的转录量降低的确认,可以通过将从该基因转录的mrna的量与非修饰菌株进行比较而进行。作为评价mrna的量的方法,可举出:northern杂交、rt-pcr、微阵列、rna-seq等(sambrook,j.,et al.,molecular cloning:a laboratory manual/third edition,cold spring harbor laboratory press,cold spring harbor(usa),2001)。mrna的量(例如,每细胞的分子数),例如,可以降低至非修饰菌株的50%以下、20%以下、10%以下、5%以下或0%。
[0266]
蛋白质的量降低的确认,可以使用抗体通过蛋白质印迹法而进行(sambrook,j.,et al.,molecular cloning:a laboratory manual/third edition,cold spring harbor laboratory press,cold spring harbor(usa),2001)。蛋白质的量(例如,每细胞的分子数),例如,可以降低至非修饰菌株的50%以下、20%以下、10%以下、5%以下或0%。
[0267]
基因破坏可以通过下述方式确认:根据破坏中使用的手段,对该基因的一部分或全部的碱基序列、制限酶图谱或全长等进行确定。
[0268]
就转化而言,例如,可以通过霉菌、酵母等的真核微生物的转化中通常使用的手法而进行。作为这样的手法,可举出原生质体法。
[0269]
<1-3>靶蛋白的制造方法
[0270]
可以利用表达诱导物质和本发明的微生物,制造靶蛋白。具体而言,可以通过在表达诱导物质的存在下对本发明的微生物进行培养,而制造靶蛋白。即,具体而言,本发明的靶蛋白的制造方法可以是包含通过用含有表达诱导物质的培养基对本发明的微生物进行培养的靶蛋白的制造方法。可以通过表达诱导物质,而对基于本发明的微生物的靶蛋白的生产进行诱导。
[0271]
使用的培养基,只要含有表达诱导物质,使得本发明的微生物能够增殖,生产靶蛋白,则没有特别限制。作为培养基,例如,可以使用除了表达诱导物质之外,还根据需要而含有选自碳源、氮源、磷酸源、硫源、其它各种有机成分、无机成分的成分的培养基。本领域技术人员可以适宜设定培养基成分的种类、浓度。对于具体的培养基组成,例如,可以参照与talaromyces cellulolyticus有关的报告(日本特开2003-135052、日本特开2008-271826、日本特开2008-271927等)中所述的培养基组成、里氏木霉等的其它各种纤维素酶生产微生物用的培养基组成。
[0272]
就碳源而言,只要能够使本发明的微生物同化并生成靶蛋白,则没有特别限制。作为碳源,例如可举出:糖类、纤维素类基质。作为糖类,具体而言,例如可举出:葡萄糖、果糖、半乳糖、木糖、阿拉伯糖、蔗糖、乳糖、纤维二糖、糖蜜、淀粉水解物、生物质水解物。作为纤维素类基质,具体而言,例如可举出:微晶纤维素(avicel)、滤纸、废纸、纸浆、木材、稻草
(ricestraw)、麦秆(straw)、稻壳、米糠、麦麸、甘蔗渣、咖啡渣、茶渣。纤维素类基质可以在进行水热分解处理、酸处理、碱处理、蒸煮、破碎、粉碎等的预处理之后用作碳源。作为市售的适宜的纤维素类基质,可举出solka floc(international fiber corp,north tonawanda,ny,u.s.a)。作为碳源,可以使用1种碳源,也可以组合使用2种或2种以上的碳源。需要说明的是,表达诱导物质本身也可以用作碳源。在表达诱导物质用作碳源的情况下,表达诱导物质可以用作唯一碳源(sole carbon source),也可以不是。表达诱导物质通常优选与其他碳源组合使用。
[0273]
作为氮源,具体而言,例如可举出:硫酸铵、氯化铵、磷酸铵等的铵盐、蛋白胨、酵母提取物、肉提取物、玉米浆、大豆蛋白分解物等的有机氮源、氨、尿素。作为氮源,可以使用1种氮源,也可以组合使用2种或2种以上的氮源。
[0274]
作为磷酸源,具体而言,例如可举出:磷酸二氢钾、磷酸氢二钾等的磷酸盐、焦磷酸等的磷酸聚合物。作为磷酸源,可以使用1种磷酸源,也可以组合使用2种或2种以上的磷酸源。
[0275]
作为硫源,具体而言,例如可举出:硫酸盐、硫代硫酸盐、亚硫酸盐等的无机硫化合物、半胱氨酸、胱氨酸、谷胱甘肽等的含硫氨基酸。作为硫源,可以使用1种硫源,也可以组合使用2种或2种以上的硫源。
[0276]
作为其它各种有机成分、无机成分,具体而言,例如可举出:氯化钠、氯化钾等的无机盐类;铁、锰、镁、钙等的微量金属类;维生素b1、维生素b2、维生素b6、烟酸、烟酸酰胺、维生素b12等的维生素类;氨基酸类;核酸类;包含这些的蛋白胨、酪蛋白氨基酸、酵母提取物、大豆蛋白分解物等的有机成分。作为其它各种有机成分、无机成分,可以使用1种成分,也可以组合使用2种或2种以上的成分。
[0277]
就培养条件而言,只要本发明的微生物能够增殖,生产靶蛋白,则没有特别限制。就培养而言,例如,可以在丝状菌等的微生物的培养中使用的通常的条件而进行。对于具体的培养条件,例如,可以参照与talaromyces cellulolyticus有关的报告(日本特开2003-135052、日本特开2008-271826、日本特开2008-271927等)所述的培养条件、里氏木霉等的其它各种纤维素酶生产微生物用的培养条件。
[0278]
就培养而言,例如,可以使用液体培养基,在好氧条件下进行。在好氧条件下的培养,具体而言,可以通过通气培养、振荡培养、搅拌培养或它们的组合而进行。就培养温度而言,例如,可以为15~43℃,特别可以为约30℃。就培养期间而言,例如,可以为2小时~20日。培养可以通过分批培养(batch culture)、流加培养(fed-batch culture)、连续培养(continuous culture)或它们的组合而实施。需要说明的是,也将培养开始时的培养基称为“初始培养基”。此外,也将流加培养或连续培养中供给至培养体系(发酵槽)中的培养基称为“流加培养基”。此外,也将在流加培养或连续培养中向培养体系中供给流加培养基称为“流加”。此外,培养可以分为预培养和主培养而实施。例如,可以在琼脂培养基等的固体培养基中进行预培养,可以在液体培养基中进行主培养。可以继续培养,例如,直至培养基中的碳源被消耗或直至本发明的微生物的活性丧失为止。
[0279]
在本发明中,各培养基成分可以含有在初始培养基、流加培养基或这两者中。初始培养基中含有的成分的种类可以与流加培养基中含有的成分的种类相同,也可以不同。此外,初始培养基中含有的各成分的浓度可以与流加培养基中含有的各成分的浓度相同,也
可以不同。此外,可以使用含有的成分的种类和/或浓度的不同的2种或2种以上的流加培养基。例如,在间歇性地进行多次流加的情况下,各回的流加培养基中含有的成分的种类和/或浓度可以相同,也可以不同。
[0280]
表达诱导物质可以在培养的整个期间中含有在培养基中,也可以仅在培养的一部分的期间中含有在培养基中。即,“通过含有表达诱导物质的培养基而培养本发明的微生物”不一定意味着表达诱导物质在培养的整个期间中含有在培养基。例如,表达诱导物质可以从培养开始时含有在培养基中,也可以不是。在表达诱导物质在培养开始时不含有在培养基中的情况下,在培养开始后将表达诱导物质供给至培养基中。供给的时机可以根据培养时间等的各种条件而适宜设定。例如,可以在菌体开始生长后将表达诱导物质供给至培养基中。供给的时机,具体而言,例如,可以在培养开始的3小时之后、6小时之后、10小时之后或20小时之后。此外,“一部分的期间”是指,例如,可以是培养的整个期间的10%以上、20%以上、30%以上、40%以上、50%以上、60%以上、70%以上、80%以上、90%以上、或95%以上的期间。表达诱导物质可以在培养中被消耗,也可以不被消耗。在任何情况下,表达诱导物质都可以追加性地供给至培养基。将表达诱导物质供给至培养基的手段没有特别限制。例如,可以通过含有表达诱导物质的流加培养基的流加,而将表达诱导物质供给至培养基。这样的流加培养基可以含有仅表达诱导物质,也可以不是。这样的流加培养基,例如,除了表达诱导物质之外,还可以含有葡萄糖等的碳源。就培养基中的表达诱导物质浓度而言,只要诱导靶蛋白的生产,则没有特别限制。就培养基中的表达诱导物质浓度而言,例如,可以为0.0005g/l以上、0.001g/l以上、0.003g/l以上、0.005g/l以上、0.01g/l以上、0.05g/l以上、0.1g/l以上、0.5g/l以上、1g/l以上、5g/l以上或10g/l以上,可以为100g/l以下、50g/l以下、10g/l以下、5g/l以下、2g/l以下、1g/l以下或0.5g/l以下,也可以是与它们不矛盾的组合。表达诱导物质可以在培养的整个期间中以所述例举的浓度范围而含有在培养基中,也可以不是。就表达诱导物质而言,例如,可以在培养开始时以所述例举的浓度范围而含有在培养基中,也可以以变成所述例举的浓度范围的方式而在培养开始后供给至培养基。在培养分为种培养和主培养而进行的情况下,靶蛋白至少在主培养的期间生产即可。即,表达诱导物质在主培养的期间(即,主培养的整个期间或主培养的一部分的期间)含有在培养基中即可,可以在种培养的期间含有在培养基中,也可以不是。这样的情况下,“培养期间(培养的期间)”、“培养的开始”等的培养中涉及的术语,可以解读为与主培养有关。
[0281]
作为表达诱导物质,可以使用市售品,也可以使用适宜制造而取得的物质。表达诱导物质的制造方法没有特别限制。就表达诱导物质而言,例如,可以通过化学合成、酶转化或它们的组合而制造。具体而言,例如,龙胆二糖等的二糖可以通过从糖原料的酶转化而制造。即,此外,本发明的靶蛋白的制造方法可以包含通过从糖原料的酶转化而制造龙胆二糖等的二糖。稍后将描述基于从糖原料的酶转化的龙胆二糖等的二糖的制造方法。
[0282]
表达诱导物质等的各种成分的浓度可以通过气相色谱法(hashimoto,k.et al.1996.biosci.biotechnol.biochem.70:22-30)、hplc(lin,j.t.et al.1998.j.chromatogr.a.808:43-49)而进行测定。
[0283]
通过如上所述对本发明的微生物进行培养,而表达靶蛋白,得到包含靶蛋白的培养物。具体而言,靶蛋白可以向培养基中、菌体表层、菌体内或它们的组合积蓄。靶蛋白特别是可以积蓄在培养基中。
[0284]
靶蛋白的生产可以通过蛋白质的检测或鉴定中使用的公知的方法而进行确认。作为这样的方法,例如可举出:sds-page、蛋白质印迹法(westernblotting)、质谱法、n末氨基酸序列分析、酶活性测定。这些方法可以单独使用1种,也可以组合使用2种或2种以上。以下,将举例表示纤维素酶的情况。就纤维素酶的生产而言,例如,可以通过测定培养物、培养上清液等的适当的部分的纤维素酶活性来确认。纤维素酶活性可以通过公知的手法而测定。具体而言,例如,可以将微晶纤维素(avicel)、滤纸等的纤维素作为基质而进行酶反应,将生成的还原糖量作为指标,算出与微晶纤维酶(avicelase)活性(avicel分解活性)、滤纸酶(fpase)活性(滤纸分解活性)等的基质对应的纤维素酶活性。还原糖量可以通过二硝基水杨酸(dns)法、somogyi nelson法等的公知的手法而测定。
[0285]
生成的靶蛋白可以适宜回收。即,本发明的靶蛋白的制造方法可以包含回收生成的靶蛋白。具体而言,就靶蛋白而言,可以作为包含靶蛋白的适当的部分而进行回收。作为这样的部分,例如可举出:培养物、培养上清液、菌体、菌体处理物(破碎物、溶解物、萃取物(无细胞萃取液)等)。就菌体而言,例如,可以以用丙烯酸酰胺、卡拉胶等的载体进行了固定化的固定化菌体的方式提供。
[0286]
此外,可以将靶蛋白分离纯化至期望的程度。靶蛋白可以以游离的状态下而提供,也可以以进行了在树脂等的固相中的固定化的固定化酶的状态下而提供。
[0287]
在将靶蛋白积蓄在培养基中的情况下,就靶蛋白而言,例如,可以在通过离心分离等从培养物中除去菌体等的固体成分后,从上清液中分离纯化。
[0288]
在靶蛋白在菌体内的情况下,就靶蛋白而言,例如,可以在对菌体进行破碎、溶解或萃取等的处理后,从处理物中分离纯化。菌体可以通过离心分离等而从培养物中回收。细胞的破碎、溶解或萃取等的处理可以通过公知的方法而进行。作为这样的方法,例如可举出:超声波破碎法、dyno-mill法、珠粒破碎、法式压制破碎(french press crushing)、溶菌酶处理。这些方法可以单独使用1种,也可以组合使用2种或2种以上。
[0289]
在靶蛋白积蓄在菌体表层的情况下,就靶蛋白而言,例如,可以在进行可溶化后,从可溶化物中分离纯化。可溶化可以通过公知的方法而进行。作为这样的方法,例如可举出:盐浓度的上升、表面活性剂的使用。这些方法可以单独使用1种,也可以组合使用2种或2种以上。
[0290]
靶蛋白的纯化(例如,从如上所述的上清液、处理物或可溶化物中的纯化)可以通过蛋白质的纯化中使用的公知的方法而进行。作为这样的方法,例如可举出:硫酸铵分级分离、离子交换色谱法、疏水色谱法、亲和色谱法、凝胶过滤色谱法、等电点沉淀。这些方法可以单独使用1种,也可以组合使用2种或2种以上。
[0291]
需要说明的是,培养物中,与靶蛋白同时,还可以生成积蓄其他酶,例如,纤维素酶、木聚糖酶、木糖苷酶(β-木糖苷酶)、阿拉伯呋喃糖苷酶(arabinofuranosidase)等的半纤维素酶。靶蛋白可以作为与这些其他酶形成的混合物而回收,也可以与这些其他酶分离而回收。
[0292]
回收的靶蛋白可以适宜制剂化。剂形没有特别限制,可以根据靶蛋白的使用用途等的各种条件而适宜设定。作为剂形,例如可举出:液剂、悬浮剂、散剂、片剂、丸剂、胶囊剂。在制剂化中,例如可以使用:赋形剂、粘合剂、崩解剂、润滑剂、稳定剂、矫味剂、矫臭剂、香料、稀释剂、表面活性剂等的药理学上容许的添加剂。
[0293]
靶蛋白的用途没有特别限制。以下,将举例表示纤维素酶的情况。纤维素酶可以在纤维素的分解中利用。例如,利用纤维素酶而使植物生物质中包含的纤维素成分糖化,从而得到含有葡萄糖的糖化液。此外,在纤维素酶具有木聚糖酶等的半纤维素酶活性的情况(例如,纤维素酶作为与半纤维素酶形成的混合物而得到的情况)下,纤维素酶也可以在半纤维素的分解中利用。例如,利用纤维素酶而使植物生物质中包含的半纤维素成分糖化,从而得到含有木糖、阿拉伯糖的糖化液。由此得到的糖化液,例如,可以作为碳源而在微生物的培养中利用。此外,在一个方式中,可以通过培养微生物,而制造l-氨基酸等的目标物质。对于纤维素酶引起的植物生物质的糖化和得到的糖化液的利用,例如,可以参照日本特开2016-131533的记载。
[0294]
<2>本发明的二糖的制造方法
[0295]
本发明的二糖的制造方法是利用二糖合成酶的二糖的制造方法。具体而言,本发明的二糖的制造方法可以是基于利用二糖合成酶并从糖原料出发的酶转化的二糖的制造方法。即,换而言之,本发明的二糖的制造方法是包含利用二糖合成酶并使糖原料转化为二糖的二糖的制造方法。具体而言,酶转化可以通过使二糖合成酶与糖原料接触而实施。即,更具体而言,本发明的二糖的制造方法可以是包含通过使二糖合成酶与糖原料接触而生成二糖的二糖的制造方法。也将通过酶转化而从糖原料生成二糖的反应称为“转化反应”。
[0296]
此处所说的“二糖”是指,葡萄糖的二糖(由2分子的葡萄糖而构成的二糖)。作为二糖,可举出β连接性的二糖。作为二糖,具体而言,可举出:龙胆二糖、纤维二糖、昆布二糖、槐糖。作为二糖,特别是,可举出龙胆二糖。在本发明中,可以制造1种二糖,也可以制造2种或2种以上的二糖。就二糖而言,例如,可以至少包含龙胆二糖。换而言之,在本发明中,例如,可以单独制造龙胆二糖,也可以制造龙胆二糖与其他1种或1种以上的二糖的组合。
[0297]
此处所说的“糖原料”是指,包含葡萄糖作为构成糖的糖,也包含葡萄糖本身。典型性地,糖原料可以仅包含葡萄糖作为构成糖。糖原料选自待制造的二糖以外的糖。作为糖原料,例如可举出:葡萄糖、纤维寡糖、纤维素。作为纤维寡糖,例如可举出:纤维二糖、纤维三糖、纤维四糖。作为纤维素,例如可举出所述的纤维素类基质。作为糖原料,可特别举出:葡萄糖、纤维二糖、纤维素。作为糖原料,可进一步特别举出葡萄糖。作为糖原料,可以使用1种糖原料,也可以组合使用2种或2种以上的糖原料。就糖原料而言,例如,可以至少包含葡萄糖。换而言之,作为糖原料,例如,可以单独使用葡萄糖,也可以组合使用葡萄糖与其他1种或1种以上的糖原料。
[0298]
<2-1>二糖合成酶及其制造
[0299]“二糖合成酶”是指,具有对从糖原料合成二糖的反应进行催化的活性的蛋白质(酶)。也将该活性称为“二糖合成活性”。也将编码二糖合成酶的基因称为“二糖合成酶基因”。就二糖合成酶而言,如果具有对从糖原料合成目的二糖的反应进行催化的活性,则没有特别限制。二糖合成酶可以具有对从糖原料合成1种二糖的反应进行催化的活性,也可以具有对从糖原料合成2种或2种以上的二糖的反应进行催化的活性。此外,二糖合成酶可以具有对从1种糖原料合成二糖的反应进行催化的活性,也可以具有对从2种或2种以上的糖原料合成二糖的反应进行催化的活性。就二糖合成酶而言,例如,可以至少具有对从糖原料合成龙胆二糖的反应进行催化的活性。此外,就二糖合成酶而言,例如,可以至少具有对从葡萄糖和/或纤维二糖合成二糖的反应进行催化的活性。就二糖合成酶而言,特别是,可以
至少具有对从葡萄糖合成二糖的反应进行催化的活性。就二糖合成酶而言,进一步特别是,可以至少具有对从葡萄糖和/或纤维二糖合成龙胆二糖的反应进行催化的活性。就二糖合成酶而言,进一步特别是,可以至少具有对从葡萄糖合成龙胆二糖的反应进行催化的活性。作为二糖合成酶,可以使用1种二糖合成酶,也可以组合使用2种或2种以上的二糖合成酶。作为二糖合成酶,可举出β-葡萄糖苷酶。作为β-葡萄糖苷酶,可举出:gh1-2蛋白、bgl3a蛋白。gh1-2蛋白、bgl3a蛋白如上所述。这些β-葡萄糖苷酶,例如,可以在talaromyces cellulolyticus等的真菌中发现。此外,作为β-葡萄糖苷酶,可举出:日本特开2010-227032、wo2004/035070中所述的那些。各种生物的二糖合成酶的氨基酸序列和编码这些基因的碱基序列,例如,可以从ncbi等的公开数据库取得。
[0300]
二糖合成酶基因和二糖合成酶可以分别例如具有所述例举的基因和蛋白质等的公知的基因和蛋白质的碱基序列和氨基酸序列。此外,二糖合成酶基因和二糖合成酶可以分别是所述例举的基因和蛋白质等的公知的基因和蛋白质的保守性变体。对于二糖合成酶基因和二糖合成酶的保守性变体,可以援用与gh1-2基因和gh1-2蛋白的保守性变体有关的记载。需要说明的是,“保持原本的功能”是指,在二糖合成酶的情况下,蛋白质的变体具有二糖合成活性。作为由某种二糖合成酶及其变体合成的二糖和基质而使用的糖原料均可以彼此相同,也可以不同。
[0301]
需要说明的是,二糖合成酶可以单独具有对从糖原料合成二糖的反应进行催化的活性,也可以在与其他酶的组合使用时具有对从糖原料合成二糖的反应进行催化的活性。因此,二糖合成酶的活性中所指的“糖原料”不限于糖原料本身,可以是糖原料通过其他酶而转化得到的物质(例如糖原料的分解物)。即,在一个方式中,“二糖合成活性”可以是对从糖原料或其分解物合成二糖的反应进行催化的活性。二糖合成酶,只要可以从糖原料制造二糖,则可以单独使用,也可以与其他酶组合使用。就二糖合成酶而言,例如,可以与其他酶组合使用,使得能够从糖原料制造二糖。具体而言,例如,在使用纤维素作为糖原料,组合使用二糖合成酶和纤维素酶的情况下,可以通过纤维素酶而使糖原料分解,通过二糖合成酶而从糖原料的分解物生成二糖。
[0302]
二糖合成酶可以通过在具有二糖合成酶基因的宿主中表达二糖合成酶基因来制造。需要说明的是,也将二糖合成酶基因的表达称为“二糖合成酶的表达”。只要具有二糖合成酶基因的宿主以可表达的方式具有二糖合成酶基因即可。具有二糖合成酶基因的宿主可以具有1拷贝的二糖合成酶基因,也可以具有2拷贝或2种以上的二糖合成酶基因。此外,具有二糖合成酶基因的宿主可以具有1种二糖合成酶基因,也可以具有2种或2种以上的二糖合成酶基因。
[0303]
此外,二糖合成酶可以通过使二糖合成酶基因在无细胞蛋白质合成系统中表达来制造。
[0304]
具有二糖合成酶基因的宿主可以是本来就具有二糖合成酶基因,也可以经过修饰,使其具有二糖合成酶基因。
[0305]
本来就具有二糖合成酶基因的宿主,可举出:如上所述生成二糖合成酶的生物,例如,talaromyces cellulolyticus等的真菌。
[0306]
作为以使得具有二糖合成酶基因的方式而经过了修饰的宿主,可举出导入了二糖合成酶基因的宿主。通过将二糖合成酶基因导入本来不具有二糖合成酶基因的宿主,而能
够使该宿主的二糖合成酶活性增大(赋予该宿主二糖合成酶活性)。
[0307]
此外,可以以使得二糖合成酶活性增大的方式修饰本来就具有二糖合成酶基因的宿主,并使用。即,具有二糖合成酶基因的宿主,例如,可以是以使得二糖合成酶活性增大的方式进行了修饰的宿主。
[0308]
就宿主而言,只要能够表达发挥功能的二糖合成酶,则没有特别限制。作为宿主,例如可举出:细菌、真菌、植物细胞、昆虫细胞和动物细胞。作为优选的宿主,可举出:细菌、真菌等的微生物。
[0309]
作为细菌,可举出:革兰氏阴性菌、革兰氏阳性细菌。作为革兰氏阴性菌,例如可举出:属于埃希菌(escherichia)属细菌、肠杆菌(enterobacter)属细菌、泛菌(pantoea)属细菌等的肠杆菌科(enterobacteriaceae)的细菌。作为革兰氏阳性细菌,可举出:芽孢杆菌(bacillus)属细菌、棒状杆菌(corynebacterium)属细菌等的棒状细菌、放线菌。作为埃希菌属细菌,例如可举出大肠杆菌(escherichia coli)。作为大肠杆菌,例如可举出:w3110菌株(atcc 27325)、mg1655菌株(atcc 47076)等的大肠杆菌k-12菌株;大肠杆菌k5菌株(atcc 23506);bl21(de3)菌株、rosetta 2(de3)plyss菌株等的大肠杆菌b菌株;和它们的衍生菌株。作为棒状细菌,例如可举出:谷氨酸棒状杆菌(corynebacterium glutamicum)、产氨棒杆菌(停滞棒状杆菌)(corynebacterium ammoniagenes(corynebacterium stationis))。
[0310]
这些菌株,例如,可以从atcc(american type culture collection)(地址:12301 parklawn drive,rockville,maryland 20852p.o.box 1549,manassas,va 20108,united states of america)订购。即,赋予与各菌株对应的登录编号,可以利用该登录编号而订购(参照http://www.atcc.org/)。与各菌株对应的登录编号记载在atcc(american type culture collection)的产品目录中。此外,例如,这些菌株可以从保藏各菌株的保藏机关入手。此外,例如,这些菌株可以从市售品入手。
[0311]
以下,对使二糖合成酶等的蛋白质的活性增大的手法(也包含导入二糖合成酶基因等的基因的方法)进行说明。
[0312]“蛋白质的活性增大”是指,与非修饰菌株相比该蛋白质的活性增大。具体而言,“蛋白质的活性增大”可以是指,与非修饰菌株相比该蛋白质在每个细胞中的活性增大。此处所说的“非修饰菌株”是指,未以使得靶蛋白的活性增大的方式而修饰的对照菌株。作为非修饰菌株,可举出:野生菌株、亲本菌株。作为非修饰菌株,具体而言,可举出宿主的说明中例举的菌株。需要说明的是,也将“蛋白质的活性增大”称为“使蛋白质的活性增强”。更具体而言,“蛋白质的活性增大”可以是指,与非修饰菌株相比,该蛋白质在每个细胞中的分子数增加,和/或,该蛋白质的每个分子的功能增大。即,“蛋白质的活性增大”这样的情况的“活性”可以不限于蛋白质的催化剂活性,还可以是指编码蛋白质的基因的转录量(mrna量)或翻译量(蛋白质的量)。此外,“蛋白质的活性增大”是指,不仅使本来就具有靶蛋白的活性的菌株中该蛋白质的活性增大,而且还包含赋予本来就不存在靶蛋白的活性的菌株该蛋白质的活性。此外,作为结果,只要蛋白质的活性增大,则可以使宿主本来具有的靶蛋白的活性降低或消失,赋予适宜的靶蛋白的活性。
[0313]
蛋白质的活性的增大的程度,只要与非修饰菌株相比蛋白质的活性增大,则没有特别限制。例如,蛋白质的活性可以上升至非修饰菌株的1.5倍以上、2倍以上或3倍以上。此外,在非修饰菌株不具有靶蛋白的活性的情况下,只要通过导入编码该蛋白质的基因而生
成该蛋白质即可,例如,可以生产该蛋白质至能够测定其酶活性的程度。
[0314]
这样的使蛋白质的活性增大的修饰,例如,可以通过使编码该蛋白质的基因的表达上升而达成。“基因的表达上升”是指,与野生菌株、亲本菌株等的非修饰菌株相比该基因的表达增大。具体而言,“基因的表达上升”可以是指,与非修饰菌株相比该基因在每个细胞中的表达量增大。更具体而言,“基因的表达上升”可以是指,基因的转录量(mrna量)增大,和/或,基因的翻译量(蛋白质的量)增大。需要说明的是,也将“基因的表达上升”称为“使基因的表达增强”。就基因的表达而言,例如,可以上升至非修饰菌株的1.5倍以上、2倍以上或3倍以上。此外,“基因的表达上升”是指,不仅使本来就表达目的基因的菌株中该基因的表达水平上升,而且包含在本来不表达目的基因的菌株中,使该基因表达。即,例如,“基因的表达上升”,包含将该基因导入不保持目的基因的菌株中,使该基因表达。
[0315]
就基因的表达的上升而言,例如,可以通过使基因的拷贝数增加而达成。
[0316]
基因的拷贝数的增加可以通过向宿主的染色体导入该基因而达成。向染色体的基因的导入,例如,可以利用同源重组而进行(miller,j.h.experiments in molecular genetics,1972,cold spring harbor laboratory)。作为利用同源重组的基因导入法,例如可举出:red-驱动整合(red-driven integration)法(datsenko,k.a,and wanner,b.l.proc.natl.acad.sci.usa.97:6640-6645(2000))等的使用直链状dna的方法、使用包含温度感受性复制起点的质粒的方法、使用可以接合转移的质粒的方法、不具有在宿主内发挥功能的复制起点的自杀载体的方法、使用噬菌体的转导(transduction)法。基因可以仅导入1拷贝,也可以导入2拷贝或其以上。例如,将在染色体上存在多份拷贝的序列作为目的而进行同源重组,从而能够向染色体导入基因的多份的拷贝。作为在染色体上存在多份拷贝的序列,可举出:重复dna序列(repetitive dna)、存在于转座子的两端的反向重复序列。此外,可以将目标物质的生产中不需要的基因等的染色体上的适当的序列作为目的而进行同源重组。此外,基因可以使用转座子、mini-mu而随机地导入至染色体上(日本特开平2-109985号公报,us5882888,ep805867b1)。
[0317]
染色体上是否导入目的基因的确认,可以通过使用了与该基因的全部或一部分具有互补性的序列的探针的southern杂交或使用了基于该基因的序列而制成的引物的pcr等来确认。
[0318]
此外,基因的拷贝数的增加可以通过将包含该基因的载体导入宿主而达成。例如,使包含目的基因的dna片段与在宿主中发挥功能的载体连接而构建该基因的表达载体,通过该表达载体而使宿主转化,从而能够使该基因的拷贝数增加。包含目的基因的dna片段,例如,可以通过将具有目的基因的微生物的基因组dna设为模板的pcr而取得。作为载体,可以使用在宿主的细胞内可以自主复制的载体。载体优选是多拷贝载体。此外,为了选择转化体,优选载体具有抗生物质抗性基因等的标记。此外,载体可以具备用于表达插入的基因的启动子、终止子。就载体而言,例如,可以是源自细菌质粒的载体、源自酵母质粒的载体、源自噬菌体的载体、粘粒或噬菌粒等。作为大肠杆菌等的肠杆菌科的细菌中可以自主复制的载体,具体而言,例如可举出:puc19、puc18、phsg299、phsg399、phsg398、pbr322、pstv29(均可从takara bio公司入手)、pacyc184、pmw219(nippon gene公司)、ptrc99a(pharmacia公司)、pprok类载体(clontech公司)、pkk 233-2(clontech公司)、pet类载体(novagen公司)、pqe类载体(qiagen公司)、pcold tf dna(takara bio公司)、pacyc类载体、广宿主域载体
rsf1010。作为在棒状细菌中可以自主复制的载体,具体而言,例如可举出:phm1519(agric.biol.chem.,48,2901-2903(1984));pam330(agric.biol.chem.,48,2901-2903(1984));具有改良了这些药剂抗性基因的质粒;pcry30(日本特开平3-210184);pcry21、pcry2ke、pcry2kx、pcry31、pcry3ke和pcry3kx(日本特开平2-72876,美国专利5185262号);pcry2和pcry3(日本特开平1-191686);paj655、paj611和paj1844(日本特开昭58-192900);pcg1(日本特开昭57-134500);pcg2(日本特开昭58-35197);pcg4和pcg11(日本特开昭57-183799);pvk7(日本特开平10-215883);pvk9(us2006-0141588);pvc7(日本特开平9-070291);pvs7(wo2013/069634)。
[0319]
在导入基因的情况下,只要基因以可表达的方式保持在宿主中即可。具体而言,只要导入基因,以使其受到基于在宿主中发挥功能的启动子序列的控制而进行表达即可。启动子可以是源自宿主的启动子,也可以是源自异源的启动子。启动子可以是待导入的基因的固有的启动子,也可以是其他基因的启动子。作为启动子,例如,如后所述,可以利用更强力的启动子。
[0320]
可以将转录终止用的终止子配置在基因的下游。终止子只要在宿主中发挥功能,则没有特别限制。终止子可以是源自宿主的终止子,也可以是源自异源的终止子。终止子可以是待导入的基因的固有的终止子,也可以是其他基因的终止子。作为终止子,具体而言,例如可举出:t7终止子、t4终止子、fd噬菌体终止子、tet终止子和trpa终止子。
[0321]
可以在各种微生物中利用的载体,与启动子、终止子有关,例如,详细地记载在“微生物学基础讲座8基因工程,共立出版,1987年”中,可以利用这些。
[0322]
此外,在导入2个或2个以上的基因的情况下,只要各基因以可表达的方式保持在宿主中即可。例如,各基因可以全部保持在单一的表达载体上,也可以全部保持在染色体上。此外,各基因可以分别保持在多个表达载体上,也可以分别保持在单一或多个表达载体上和染色体上。此外,可以通过2个或2个以上的基因而构成操纵子并导入。作为“导入2或2个以上的基因的情况”,例如可举出:导入分别编码2或2个以上的蛋白质的基因的情况、导入分别编码构成单一的蛋白质复合体的2或2个以上的亚基的基因的情况、和它们的组合。
[0323]
导入的基因,只要编码在宿主中发挥功能的蛋白质,则没有特别限制。导入的基因可以是源自宿主的基因,也可以是源自异源的基因。就导入的基因而言,例如,可以使用基于该基因的碱基序列而设计的引物,将具有该基因的生物的基因组dna、搭载该基因的质粒等作为模板,通过pcr而取得。此外,例如,导入的基因可以基于该基因的碱基序列而进行全合成(gene,60(1),115-127(1987))。取得的基因可以原样利用或适宜修饰后利用。即,通过对基因进行修饰,而取得该基因的变体。基因的修饰可以通过公知的手法而进行。例如,可以通过位点特异性突变法,而在dna的目的位点导入目的变异。即,例如,可以通过位点特异性突变法,而对基因的编码区域进行修饰,以使得编码的蛋白质在特定的位点中包含氨基酸残基的取代、缺失、插入和/或添加。作为位点特异性突变法,可举出:使用pcr的方法(higuchi,r.,61,in pcr technology,erlich,h.a.eds.,stockton press(1989);carter,p.,meth.in enzymol.,154,382(1987))、使用噬菌体的方法(kramer,w.and frits,h.j.,meth.in enzymol.,154,350(1987);kunkel,t.a.et al.,meth.in enzymol.,154,367(1987))。此外,可以全合成基因的变体。
[0324]
需要说明的是,当蛋白质作为包含多个亚基的复合体而发挥功能的情况下,作为
结果,只要蛋白质的活性增大,则可以对所有的这些多个亚基进行修饰,也可以仅对一部分进行修饰。即,例如,在通过使基因的表达上升而使蛋白质的活性增大的情况下,可以使编码这些亚基的多个基因的所有的表达均增强,也可以仅使一部分的表达增强。通常,优选使编码这些亚基的多个基因的所有的表达增强。此外,就构成复合体的各亚基而言,只要复合体具有靶蛋白的功能,则可以源自1种生物,也可以源自2种或2种以上的不同的生物。即,例如,编码多个亚基,可以将源自相同的生物的基因导入宿主,也可以将分别源自不同的生物的基因导入宿主。
[0325]
此外,基因的表达的上升可以通过使基因的转录效率提高而达成。此外,基因的表达的上升可以通过使基因的翻译效率提高而达成。基因的转录效率、翻译效率的提高例如可以通过表达调控序列的修饰而达成。“表达调控序列”是影响基因的表达的位点的总称。作为表达调控序列,例如可举出:启动子、shine-dalgarno(sd)序列(也称为核糖体结合位点(rbs))、和rbs与起始密码子之间的间隔(spacer)区域。表达调控序列可以使用启动子检索载体、genetyx等的基因分析软件而进行确定。这些表达调控序列的修饰例如可以通过使用了温度感受性载体的方法、red-驱动整合法(wo2005/010175)而进行。
[0326]
就基因的转录效率的提高而言,例如,可以通过用更强力的启动子取代染色体上的基因的启动子而达成。“更强力的启动子”是指,基因的转录比原本存在的野生型的启动子更高的启动子。作为更强力的启动子,例如可举出:作为公知的高表达启动子的t7启动子、trp启动子、lac启动子、thr启动子、tac启动子、trc启动子、tet启动子、arabad启动子、rpoh启动子、msra启动子、源自bifidobacterium的pm1启动子、pr启动子和pl启动子。此外,作为能够在棒状细菌中利用的更强力的启动子,可举出:人为性地设计变更的p54-6启动子(appl.microbiol.biotechnol.,53,674-679(2000))、可以在棒状细菌内通过乙酸、乙醇、丙酮酸等诱导的pta、acea、aceb、adh、amye启动子、作为在棒状细菌内表达量多的强力的启动子的cspb、sod、tuf(ef-tu)启动子(journal of biotechnology 104(2003)311-323,appl environ microbiol.2005dec;71(12):8587-96.)、lac启动子、tac启动子、trc启动子。此外,作为更强力的启动子,可以通过使用各种报告基因,而取得天然启动子的高活性型的启动子。例如,通过使启动子区域内的-35、-10区域接近共有序列,而能够提高启动子的活性(国际公开第00/18935号)。作为高活性型启动子,可举出:各种tac样启动子(katashkina ji et al.russian federation patent application 2006134574)、pnlp8启动子(wo2010/027045)。启动子的强度的评价法和强力的启动子的实例记载在goldstein等人的论文(prokaryotic promotersin biotechnology.biotechnol.annu.rev.,1,105-128(1995))等中。
[0327]
就基因的翻译效率的提高而言,例如,可以通过用更强力的sd序列取代染色体上的基因的shine-dalgarno(sd)序列(也称为核糖体结合位点(rbs))而达成。“更强力的sd序列”是指,mrna的翻译比原本存在的野生型的sd序列更高的sd序列。作为更强力的sd序列,例如可举出源自噬菌体t7的基因10的rbs(olins p.o.et al,gene,1988,73,227-235)。此外,已知在rbs与起始密码子之间的间隔区域,特别是在紧邻起始密码子的上游的序列(5
’-
utr)中的几个核苷酸的取代、或插入、或缺失对mrna的稳定性和翻译效率具有显著影响,能够通过对它们进行修饰而提高基因的翻译效率。
[0328]
就基因的翻译效率的提高而言,例如,可以通过密码子的修饰而达成。例如,通过
用利用频率更高的同义密码子取代存在于基因中的稀有密码子,而能够提高基因的翻译效率。即,例如,可以修饰导入的基因,使得根据使用的宿主的密码子使用频率而具有最适的密码子。就密码子的取代而言,例如,可以通过在dna的目的位点导入目的变异的位点特异性突变法而进行。此外,可以全合成密码子被取代的基因片段。各种的生物中的密码子的使用频率在“密码子使用数据库”(http://www.kazusa.or.jp/codon;nakamura,y.et al,nucl.acids res.,28,292(2000))中公开。
[0329]
此外,基因的表达的上升可以通过下述方式达成:以使得基因的表达上升的方式使调节子扩增,或,以使得基因的表达降低的方式使调节子缺失或弱化。
[0330]
如上所述的使基因的表达上升的手法可以单独使用,也可以任意的组合使用。
[0331]
此外,这样的使蛋白质的活性增大的修饰,例如,可以通过使蛋白质的比活性增强而达成。比活性的增强也包含反馈抑制的降低和解除。就比活性增强的蛋白质而言,例如,可以探索各种的生物而取得。此外,可以通过在天然的蛋白质中导入变异而取得高活性型的蛋白质。就导入的变异而言,例如,可以是蛋白质的1或者几个位置的1个或几个氨基酸被取代、缺失、插入或添加。例如,变异的导入可以通过如上所述的位点特异性突变法而进行。此外,例如,变异的导入可以通过突变处理而进行。作为突变处理,可举出:x射线的照射、紫外线的照射、以及基于n-甲基-n'-硝基-n-亚硝基胍(mnng)、甲磺酸乙酯(ems)和甲基磺酸甲酯(mms)等的变异剂的处理。此外,可以在体外(in vitro)dna直接用羟胺进行处理,诱发随机变异。比活性的增强可以单独使用,也可以与如上所述的使基因的表达增强的手法任意组合而使用。
[0332]
转化的方法没有特别限定,可以使用以往已知的方法。例如可以使用:针对大肠杆菌k-12而报告的用氯化钙对受体菌细胞进行处理而使dna的透过性增强的方法(mandel,m.and higa,a.,j.mol.biol.1970,53,159-162)、针对枯草芽孢杆菌而报告的从增殖阶段的细胞制备感受态细胞并导入dna的方法(duncan,c.h.,wilson,g.a.and young,f.e..,1997.gene 1:153-167)。或者,也可以应用已知针对枯草芽孢杆菌、放线菌类和酵母的、使dna受体菌的细胞在易于组入重组dna的原生质体或原生质球的状态下而将重组dna导入dna受体菌的方法(chang,s.and choen,s.n.,1979.mol.gen.genet.168:111-115;bibb,m.j.,ward,j.m.and hopwood,o.a.1978.nature 274:398-400;hinnen,a.,hicks,j.b.and fink,g.r.1978.proc.natl.acad.sci.usa 75:1929-1933)。或者,可以利用针对棒状细菌而报告的电脉冲法(日本特开平2-207791)。
[0333]
可以通过测定该蛋白质的活性来确认蛋白质的活性增大。
[0334]
可以通过确认编码该蛋白质的基因的表达上升来确认蛋白质的活性增大。基因的表达上升可以通过下述方式进行确认:确认该基因的转录量上升、确认从该基因表达的蛋白质的量上升。
[0335]
基因的转录量上升的确认,可以通过将从该基因转录的mrna的量与野生菌株或亲本菌株等的非修饰菌株进行比较而进行。作为评价mrna的量的方法,可举出:northern杂交、rt-pcr、微阵列、rna-seq等(sambrook,j.,et al.,molecular cloning:a laboratory manual/third edition,cold spring harbor laboratory press,cold spring harbor(usa),2001)。mrna的量(例如,每细胞的分子数),例如,可以上升至非修饰菌株的1.5倍以上、2倍以上或3倍以上。
[0336]
蛋白质的量上升的确认,可以使用抗体通过蛋白质印迹法而进行(molecular cloning(cold spring harbor laboratory press,cold spring harbor(usa),2001))。蛋白质的量(例如,每细胞的分子数),例如,可以上升至非修饰菌株的1.5倍以上、2倍以上或3倍以上。
[0337]
所述的使蛋白质的活性增大的手法不限于二糖合成酶的活性增强、二糖合成酶基因的导入,可以利用在任意的蛋白质的活性增强中、任意的基因,例如编码这些任意的蛋白质的基因的表达增强中。
[0338]
通过在培养基中培养具有二糖合成酶基因的宿主,而能够表达二糖合成酶。此时,根据需要,可以进行二糖合成酶基因的表达诱导。基因的表达诱导的条件可以根据基因的表达系统的构成等的各种条件而适宜选择。
[0339]
就培养基组成、培养条件而言,只要具有二糖合成酶基因的宿主能够增殖,生产二糖合成酶,则没有特别限制。培养基组成、培养条件可以根据宿主的种类等的各种条件而适宜设定。就培养而言,例如,可以使用培养细菌、真菌等的微生物利用的通常的培养基并在通常的条件下实施。对于用于培养细菌、培养的具体的培养基组成、培养条件,例如,可以参照在利用了大肠杆菌(e.coli)、棒状细菌等的细菌的各种物质生产中利用的培养基组成、培养条件。此外,对于用于培养真菌的具体的培养基组成、培养条件,例如可以参照:与talaromyces cellulolyticus有关的报告(日本特开2003-135052、日本特开2008-271826、日本特开2008-271927等)中所述的培养基组成、培养条件;或在里氏木霉(trichoderma reesei)等的其它各种纤维素酶生产微生物的培养中利用的培养基组成、培养条件。此外,对于培养基组成、培养条件,例如,除了培养基可以含有表达诱导物质之外,还可以参照本发明的靶蛋白的制造方法中的培养基组成、培养条件。
[0340]
作为培养基,例如可以使用根据需要而含有选自碳源、氮源、磷酸源、硫源、其它各种有机成分、无机成分的成分的培养基。本领域技术人员可以适宜设定培养基成分的种类、浓度。就碳源而言,只要具有二糖合成酶基因的宿主能够利用,则没有特别限定。作为碳源,例如可举出:糖类、纤维素类基质等的所述的那些。此外,作为碳源,例如也可举出:乙酸、富马酸、柠檬酸、琥珀酸、苹果酸等的有机酸类、甘油、粗制甘油、乙醇等的醇类、脂肪酸类。作为其它培养基成分,可举出所述的那些。
[0341]
就培养而言,例如,可以使用液体培养基,在好氧条件下进行。好氧条件是指,液体培养基中的溶存氧浓度为作为基于氧膜电极的检测限界的0.33ppm以上,可以优选为1.5ppm以上。就氧浓度而言,例如,可以控制在饱和氧浓度的5~50%,优选为在10%左右。具体而言,好氧条件下的培养可以通过通气培养、振荡培养、搅拌培养或它们的组合而进行。就培养基的ph而言,例如,可以为ph3~10,优选为ph5~8。在培养中,可以根据需要对培养基的ph进行调整。培养基的ph可以使用:氨气、氨水、碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、碳酸镁、氢氧化钠、氢氧化钙、氢氧化镁等的各种碱性或酸性物质而进行调整。就培养温度而言,例如,可以为20~40℃,优选为25℃~37℃。就培养期间而言,例如,可以为10小时~120小时。培养可以通过分批培养(batch culture)、流加培养(fed-batch culture)、连续培养(continuous culture)或它们的组合而实施。可以继续培养,例如,直至培养基中的碳源被消耗或宿主的活性丧失为止。
[0342]
通过如上所述培养具有二糖合成酶基因的宿主,表达二糖合成酶,得到包含二糖
合成酶的培养物。就二糖合成酶而言,例如,可以积蓄在宿主的菌体内。“菌体”可以根据宿主的种类,适宜解读为“细胞”。需要说明的是,根据使用的宿主和二糖合成酶基因的设计,可能使二糖合成酶积蓄在周质中或使二糖合成酶分泌生产在菌体外(例如菌体表层、培养基中)。
[0343]
二糖合成酶可以以在二糖的制造可以使用的任意的形态而使用。具体而言,二糖合成酶可以以能够作用于糖原料的任意的形态而使用。例如,二糖合成酶可以以分离至期望的程度的形态而使用,也可以以含有在原材料的形态而使用。换而言之,“二糖合成酶”可以是指纯化至期望的程度的二糖合成酶(纯化酶),也可以是指含有二糖合成酶的原材料。
[0344]
含有二糖合成酶的原材料,只要以能够将二糖合成酶作用于糖原料的方式而含有二糖合成酶,则没有特别限制。作为含有二糖合成酶的原材料,可举出:含有二糖合成酶的培养物、培养上清液、菌体、菌体处理物(破碎物、溶解物、萃取物(无细胞萃取液))。作为含有二糖合成酶的原材料,特别是,可举出含有二糖合成酶的菌体。作为含有二糖合成酶的原材料,可进一步特别举出含有二糖合成酶的大肠杆菌(e.coli)菌体。就菌体而言,例如,可以原样(原样包含在培养物中)或从培养物中回收后,在二糖的制造中使用。就菌体而言,例如,可以通过离心分离而从培养物中回收。此外,就培养物、从这些中回收的菌体而言,例如,可以适宜进行清洗、浓缩、稀释等的处理后,在二糖的制造中使用。因此,就菌体而言,例如,可以以分离至期望的程度的形态而使用,也可以以含有在原材料中的形态而使用。此外,就菌体而言,例如,可以以用丙烯酸酰胺、卡拉胶等的载体进行了固定化的固定化菌体的形态而使用。此外,二糖合成酶可以被分离纯化至期望的程度后使用。对于二糖合成酶的分离纯化,可以援用对靶蛋白的分离纯化的记载。二糖合成酶可以以游离的状态而利用,也可以以进行了在树脂等的固相中的固定化的固定化酶的状态而使用。
[0345]
如上所述形态的二糖合成酶可以使用1种形态,也可以组合使用2种或2种以上的形态。
[0346]
<2-2>二糖的制造方法
[0347]
利用二糖合成酶而实施转化反应,从而能够制造二糖。
[0348]
转化反应可以在适当的液体中实施。也将实施转化反应的液体称为“反应液”。具体而言,转化反应可以通过在适当的反应液中使二糖合成酶(例如,含有二糖合成酶的大肠杆菌(e.coli)菌体等的所述例举的形态)与糖原料共存而实施。例如,转化反应可以以分批式而实施,也可以以柱式而实施。在分批式的情况下,例如,可以通过在反应容器内的反应液中使二糖合成酶与糖原料混合,而实施转化反应。转化反应可以静置实施,也可以在搅拌、振荡下实施。在柱式的情况下,例如,可以通过使含有糖原料的反应液通过填充有固定化酶、固定化菌体的柱,而实施转化反应。作为反应液,可举出:水性缓冲液等的水性介质(水性溶剂)。
[0349]
就反应液而言,除了糖原料之外,可以根据需要含有糖原料以外的成分。作为糖原料以外的成分,可举出:ph缓冲剂、培养基成分。反应液中含有的成分的种类、浓度可以根据二糖合成酶的种类、使用方式等的各种条件而适宜设定。
[0350]
就转化反应的条件(反应液的ph、反应温度、反应时间、各种成分的浓度等)而言,只要生成二糖,则没有特别限制。例如,转化反应可以通过利用了酶、菌体的物质转化中使用的通常的条件而进行。转化反应的条件可以根据二糖合成酶的种类、使用方式等的各种
cellulolyticus等的纤维素酶生产微生物带来的纤维素酶生产的诱导中使用。此外,就二糖而言,例如,可以在本发明的靶蛋白的制造方法中使用。
[0360]
[实施例]
[0361]
以下,通过实施例而进一步对本发明进行具体性说明,但本发明的技术的范围不限于该实施例。
[0362]
(1)t.cellulolyticus的纤维素酶生产中必不可少的基因gh1-2的鉴定
[0363]
本申请发明人等发现了编码预测为glucoside hydrolase family 1(gh1)型的beta-glucosidase的蛋白质的基因作为深度参与t.cellulolyticus的纤维素酶生产的基因。以下,将该基因记为“gh1-2”。此外,将gh1-2基因所编码的蛋白质记为“gh1-2”。
[0364]
(1-1)t.cellulolyticus f09δgh1-2菌株与f09pyrf+菌株的制备
[0365]
将t.cellulolyticus f09菌株(日本特开2016-131533)作为亲本菌株,通过以下步骤,而破坏gh1-2基因(seq id no.1),制备f09δgh1-2菌株。f09菌株是在将t.cellulolyticus s6-25菌株(nite bp-01685)作为亲本菌株而得到的在pyrf基因中具有变异(一碱基取代)的菌株。f09菌株通过pyrf基因的变异而表现出尿嘧啶(uracil)缺陷型。
[0366]
首先,根据以下的步骤而制成:具有按照t.cellulolyticus的gh1-2基因上游区域、pyrf基因标记、gh1-2基因下游区域的顺序连接而成的碱基序列的gh1-2破坏用dna片段。
[0367]
将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.2和3)的pcr而使gh1-2基因的上游区域扩增,通过使用了引物(seq id no.4和5)的pcr而使gh1-2基因的下游区域扩增。此外,将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.6和7)的pcr而使pyrf基因的整个区域(包含启动子和终止子)扩增。使用wizard sv gel and pcr clean-up system(promega)而分别使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了gh1-2破坏用dna片段的puc-gh1-2::pyrf质粒。将puc-gh1-2::pyrf质粒作为模板,通过使用了引物(seq id no.2和5)的pcr而使gh1-2破坏用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0368]
接下来,将f09菌株接种至包含12g/l potato dextrose broth(difco)、20g/l bacto agar(difco)的培养基(以下,称为“pd培养基”)中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的gh1-2破坏用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置
15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种在包含1m蔗糖的最小必需培养基(表1)中,在30℃下培养7天,从而选拔互补了尿嘧啶(uracil)缺陷型的菌株。将出现的菌落接种至最小必需培养基中,在30℃下培养4天后,确认gh1-2基因被pyrf基因取代,得到f09δgh1-2菌株。
[0369]
[表1]
[0370]
表1
[0371][0372][0373]
此外,作为控制菌株,通过以下步骤,使f09菌株pyrf基因得到互补,制备互补了尿嘧啶(uracil)缺陷型的f09pyr+菌株。
[0374]
将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.8和9)的pcr而使pyrf基因的整个区域(包含启动子和终止子)扩增。使用wizard sv gel and pcr clean-up system(promega)而使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了pyrf基因的整个区域(包含启动子和终止子)的puc-pyrf质粒。将puc-pyrf质粒作为模板,通过使用了引物(seq id no.7和8)的pcr而使pyrf基因互补用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0375]
接下来,将f09菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)
30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的pyrf基因互补用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖的最小必需培养基中,在30℃下培养7天,从而选拔互补了尿嘧啶(uracil)缺陷型的菌株。将出现的菌落接种至最小必需培养基中,在30℃下培养4天后,确认变异型的pyrf基因被野生型的pyrf基因取代,得到f09pyr+菌株。
[0376]
(1-2)将纤维素设为碳源的烧瓶培养上清液样品的制备
[0377]
通过将作为纤维素基质的solka-floc(international fiber公司)设为碳源的烧瓶培养而对基于t.cellulolyticus f09δgh1-2菌株和f09pyr+菌株的纤维素酶生产进行评价。
[0378]
<烧瓶培养>
[0379]
分别将f09δgh1-2菌株和f09pyr+菌株接种至1/2pda平板(表2)中,在30℃下培养3天。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘,接种至sf烧瓶培养基(表2)中,在30℃、220rpm的条件下进行7天旋转培养。进行适宜取样,通过0.22μm的针头过滤器而除去菌体,将得到的培养上清液作为酶液。
[0380]
[表2]
[0381]
表2
[0382]
<sf烧瓶培养基组成>
[0383][0384][0385]
※
ph4.0(h2so4)
[0386]
<1/2pda平板组成>
[0387][0388]
(1-3)烧瓶培养上清液样品的电泳
[0389]
对样品溶液进行sds-page和cbb染色。具体而言,使用any kd
tm mini-protean(注册商标)tgx
tm precast protein gels(bio-rad)和powerpac
tm basic power supply(bio-rad)而进行泳动,通过bio-safe
tm coomassie stain(bio-rad)而进行染色。
[0390]
将结果示于图1。在作为控制菌株的f09pyr+菌株的情况下,观察到推测为纤维素酶的蛋白质的分泌,而在f09δgh1-2菌株的情况下,几乎没有观察到蛋白质的分泌(图1左)。此外,当将培养液静置24小时并进行观察时,与f09pyr+菌株不同,在f09δgh1-2菌株的情况下,观察到作为基质添加的固体成分的solka-floc的沉淀(图1右)。这些结果显示,与f09pyr+菌株相比,在f09δgh1-2菌株的情况下,纤维素酶的分泌生产能力和纤维素的同化能力显著降低。
[0391]
(1-4)t.cellulolyticus f09δgh1-2菌株在各种碳源中的生长和分泌纤维素酶的晕圈测定
[0392]
由于显示在f09δgh1-2菌株的情况下,纤维素酶的分泌生产能力和纤维素的同化能力显著降低,因此,对使用各种碳源时的生长和纤维素酶的分泌生产进行评价。步骤如下所示。
[0393]
分别将f09δgh1-2菌株和f09pyr+菌株接种至最小必需培养基中,在30℃下进行3天培养。使用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘分别静置在包含各种碳源的最小必需培养基和pd培养基中,在30℃进行培养并观察生长。各种碳源为10g/l的葡萄糖、纤维二糖、solka-floc(international fiber公司)和羧甲基纤维素(平均mw约250000,sigma,以下称为“cmc”)。
[0394]
将结果示于图2。在将葡萄糖设为碳源的最小必需培养基和pd培养基中,在f09pyr+菌株和f09δgh1-2菌株的情况下未观察到生长差异。另一方面,在将作为纤维素基质的纤维二糖、solka-floc或cmc设为碳源的最小必需培养基中,与f09pyr+菌株相比,观察到f09δgh1-2菌株中菌落生长的降低、菌丝的粗密化。
[0395]
此外,就包含cmc的琼脂培养基而言,可以利用通过纤维素酶而一部分得到分解的cmc不易通过congo-red而染色,在评价分泌纤维素酶的晕圈测定中使用。分别使f09δgh1-2菌株和f09pyr+菌株通过与所述同样的步骤而在将cmc设为碳源的最小必需培养基上生长。培养后,添加以10:1混合50mm磷酸钠缓冲液(ph 7.0)与2mg/ml的congo red(nacalai tesque)而得到的congo red溶液,使其涂覆平板表面,在室温下静置30分钟。然后,除去平板上的congo red溶液,添加磷酸钠缓冲液(50mm,ph 7.0),使其涂覆平板表面。再次,在室温下静置30分钟后,除去平板上的磷酸钠缓冲液,稍稍使平板表面干燥,对晕圈进行观察。
[0396]
结果示于图2。在f09pyr+菌株中,在菌落外部观察到晕圈,而在f09δgh1-2菌株中,在菌落外部未观察到晕圈。
[0397]
这些结果显示,gh1-2基因编码的被预测为gh1型的beta-glucosidase的蛋白质(gh1-2)是t.cellulolyticus的纤维素酶的分泌生产和纤维素的同化中重要的基因。
[0398]
(2)gh1-2基因编码的蛋白质的表达和功能分析
[0399]
(2-1)gh1-2基因的cdna的取得和在大肠杆菌(e.coli)中的表达
[0400]
使用rneasy plant mini kit(qiagen)而从t.cellulolyticus y-94菌株的菌体制备rna溶液,使用smarter(注册商标)race 5
’
/3
’
kit而从该rna溶液制备全长cdna溶液。
[0401]
使用该cdna溶液和设计在gh1-2基因区域内部的5
’
/3
’
race分析用的引物(seq id no.10和seq id no.11),按照试剂盒的说明书,对转录产物的5
’
端和3
’
端进行分析。其结果,gh1-2基因的转录起始点集中在起始密码子的上游90~60碱基的区域,转录终止点分散在终止密码子的下游60~180碱基的区域。此处所说的起始密码子和终始密码子是seq id no.22的cdna序列(后述)的起始密码子和终止密码子。在gh1-2基因的全长cdna中,seq id no.22的cdna序列的外侧找不到适当的起始密码子和终止密码子的候补。
[0402]
接下来,将所述的cdna溶液作为模板,通过使用了引物(seq id no.12和13)的pcr而使gh1-2基因的cdna片段扩增。此外,将pet24a质粒(novagen)作为模板,通过使用了引物(seq id no.14和15)的pcr而使pet24a扩增。使用wizard sv gel and pcr clean-up system(promega)而分别使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而连接纯化的pcr产物。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含50mg/l卡那霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了gh1-2基因的cdna片段的pet24a-gh1-2-his6质粒。如果根据pet24a-gh1-2-his6质粒,则gh1-2以c末端添加有his标签的方式而进行表达。使用引物(seq id no.16、17、18、19、20、21)进行pet24a-gh1-2-his6质粒的序列分析,确定gh1-2基因的cdna序列(gh1-2基因的编码区域的cdna序列;seq id no.22)和由这些编码的gh1-2的氨基酸序列(seq id no.23)。该氨基酸序列不包含分泌信号序列,因此,暗示gh1-2是细胞内定位的蛋白质。
[0403]
将pet24a-gh1-2-his6质粒转化为大肠杆菌(e.coli)rosetta
tm 2(de3)plyss competent cells(novagen),在lb琼脂培养基(包含50mg/l卡那霉素)中,在37℃下培养一晚,从而形成菌落。将形成的菌落在lb琼脂培养基(包含50mg/l卡那霉素)上划线,取得单个菌落,从而得到ecghh6菌株。
[0404]
将大肠杆菌(e.coli)ecghh6菌株接种至试验管内的3ml的包含50mg/l卡那霉素的lb培养基中,在37℃、120rpm的条件下振荡一晚而进行培养。将培养液以1/100的量接种至50ml的圆底烧瓶内的包含50mg/l卡那霉素的lb培养基中,在37℃、120rpm的条件下进行振荡培养,直至od600变为0.8。接下来,将圆底烧瓶转移至13℃的箱式振荡器内,在13℃、120rpm的条件下进行1小时振荡培养。然后,添加iptg使其变为1mm,进一步培养17小时。培养后,在4℃、5000rpm的条件下进行5min离心而回收菌体,在-20℃的冷柜中冻结1小时。然后,使用ni-nta fast start kit(qiagen)而在非改性条件下从菌体中萃取可溶性蛋白质,使用试剂盒中随附的ni-nta亲和柱、清洗缓冲液和溶出缓冲液而使c末端添加有his标签的gh1-2纯化。包含纯化gh1-2的溶出部分使用amicon ultra-15 30kda cut off(merck millipore)超滤膜过滤器和50mm磷酸钠缓冲液(ph=6.5)而进行脱盐和浓缩。然后,进行使用了sds-page、pierce fast western blot kit(thermo fisher scientific)和his标签抗体anti-his-tag mab-hrp-direct(mlb)的western分析。确认纯化的gh1-2是单条带(图3),用于之后的实验。
[0405]
(2-2)基于gh1-2的从高浓度葡萄糖的反应生成物的鉴定
[0406]
将纯化的gh1-2添加至40%葡萄糖水溶液中,使其变为1.4g/l,在40℃下进行24小时反应,得到样品。将该样品用离子交换水稀释1000倍而进行离子交换色谱法,分析反应生成物。将离子交换色谱法的条件示于表3。其结果,确认了gh1-2引起的龙胆二糖等的β连接性的葡萄糖寡糖类的生成(图4)。
[0407]
[表3]
[0408]
表3
[0409][0410][0411][0412]
由此,显示通过使用gh1-2,而能够从高浓度的葡萄糖生成龙胆二糖等的β连接性的葡萄糖寡糖类。即,显示gh1-2具有糖转移活性。
[0413]
(3)t.cellulolyticus中的β结合性葡萄糖寡糖的纤维素酶生产诱导能力的评价
[0414]
(3-1)基于各种β结合性葡萄糖寡糖的分泌纤维素酶的晕圈测定
[0415]
对通过gh1-2而生成的β连接性的葡萄糖寡糖的t.cellulolyticus中的纤维素酶生产诱导能力进行验证。步骤如下所示。
[0416]
分别添加各种β连接性的葡萄糖寡糖(纤维三糖、纤维二糖、龙胆二糖、昆布二糖、槐糖)并使浓度变为1mm,制备将cmc设为碳源的最小必需培养基(cmc最小必需培养基)。在将葡萄糖设为碳源的最小必需培养基中分别使f09pyr+菌株和f09δgh1-2菌株生长3天。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘静置在添加了所述的各种β连接性的葡萄糖寡糖的cmc最小必需培养基中,在30℃下生长36小时。培养后,添加以10:1混合50mm磷酸钠缓冲液(ph 7.0)与2mg/ml的congo red(nacalai tesque)而得到的congo red溶液,使其涂覆平板表面,在室温下静置30分钟。然后,除去平板上的congo red溶液,添加磷酸钠缓冲液(50mm,ph 7.0),使其涂覆平板表面。再次,在室温下静置30分钟后,除去平板上的磷酸钠缓冲液,稍稍使平板表面干燥,对晕圈进行观察。
[0417]
结果示于图5。显示在t.cellulolyticus中,龙胆二糖显著互补了gh1-2基因的损失引起的纤维素酶的分泌能力的降低。特别是,就龙胆二糖而言,即使其浓度为1/100量的0.01mm,也比其他β结合性葡萄糖寡糖类更强地互补了纤维素酶的分泌能力的降低。另一方面显示,通常作为纤维素酶分泌的诱导剂的纤维二糖几乎不能互补纤维素酶的分泌能力的降低。从这些结果可知,在t.cellulolyticus中,龙胆二糖表现出强纤维素酶生产诱导能力。
[0418]
(3-2)基于gh1-2的从纤维二糖的反应生成物的鉴定
[0419]
由于纤维二糖不能互补gh1-2基因的损失引起的纤维素酶分泌能力的明显的降低,因此,探讨被预测为beta-glucosidase的gh1-2在纤维二糖中发挥作用,从而参与纤维素酶分泌的可能性。
[0420]
将纯化的gh1-2添加至2%的纤维二糖水溶液中并使其变为30μg/ml,将在40℃下进行了1~60分钟反应的反应液设为样品溶液。将该样品溶液以每3μl点样在具有层厚为200μm,20
×
20cm尺寸,平均细孔径为60埃(angstrom)的二氧化硅层的薄层色谱板(merck)上并展开。展开溶剂的组成以体积比计为氯仿:甲醇:纯水=30:20:5。显色检测使用检测还原基团的苯胺邻苯二甲酸酯法。展开后使展开溶剂充分蒸发后,通过玻璃喷雾而喷涂在水饱和丁醇中溶解有苯二甲酸和苯胺的显色液,使用电热板在150℃以上进行加热,直至得到充分显色。其结果确认了,在gh1-2和纤维二糖的反应生成物中,不仅包含作为水解活性产生的生成物的葡萄糖,而且包含作为β连接性的葡萄糖寡糖的纤维三糖、龙胆二糖、槐糖和昆布二糖(图6)。因此,再次显示gh1-2不仅具有水解活性,而且具有糖转移活性。
[0421]
此外,将该样品溶液用离子交换水稀释10倍,在实施例(2-2)所述的条件下进行离子交换色谱法,分析反应生成物。其结果再次确认了,gh1-2引起的龙胆二糖等的β连接性的葡萄糖寡糖类的生成(图7)。
[0422]
这些结果暗示,t.cellulolyticus的gh1-2可以通过糖转移而在细胞内从纤维二糖生成包含龙胆二糖的β连接性的葡萄糖寡糖类。
[0423]
(4)使用了龙胆二糖的纤维素酶生产培养评价
[0424]
添加龙胆二糖而进行t.cellulolyticus的液体培养,验证纤维素酶生产中的效果。步骤如下所示。
[0425]
(4-1)t.cellulolyticus f09δsc菌株的制备
[0426]
将t.cellulolyticus f09菌株(日本特开2016-131533)作为亲本菌株,通过以下步骤,破坏sc基因,制备f09δsc菌株。sc基因编码硫酸盐同化路径的硫酸盐渗透(sulfate permease)。当使sc基因损失时,虽然变成蛋氨酸缺陷型的,但是获得对硒酸的抗性。
[0427]
首先,根据以下的步骤制成具有按照t.cellulolyticus的sc基因上游区域、pyrf基因标记、sc基因下游区域的顺序连接的碱基序列的sc破坏用dna片段。
[0428]
将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.24和25)的pcr而使sc基因的上游区域扩增,通过使用了引物(seq id no.26和27)的pcr而使sc基因的下游区域扩增。使用wizard sv gel and pcr clean-up system(promega)而分别使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了sc破坏用dna片段的puc-dsc质粒。将puc-dsc质粒作为模板,通过使用了引物(seq id no.24和27)的pcr而使sc基因破坏用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0429]
接下来,将f09菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的sc基因破坏用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖、1mm硒酸、30mg/l l-蛋氨酸、1g/l尿苷、1g/l尿嘧啶的最小必需培养基中,在30℃下培养14天,从而选拔对硒酸具有抗性的菌株。将出现的菌落接种至包含30mg/l l-蛋氨酸、1g/l尿苷、1g/l尿嘧啶的最小必需培养基中,在30℃下培养4天后,确认表现出蛋氨酸缺陷型和sc基因损失,取得f09δsc菌株。
[0430]
(4-2)t.cellulolyticus f09δgh1-2δsc菌株的制备
[0431]
将t.cellulolyticus f09δsc菌株作为亲本菌株,通过以下步骤,破坏gh1-2基因,制备f09δgh1-2δsc菌株。
[0432]
将puc-gh1-2::pyrf质粒作为模板,通过使用了引物(seq id no.2和5)的pcr而使gh1-2破坏用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0433]
接下来,将f09δsc菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻
璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的gh1-2破坏用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖、30mg/l l-蛋氨酸的最小必需培养基中,在30℃下培养7天,从而选拔互补了尿嘧啶(uracil)缺陷型的菌株。将出现的菌落接种至包含30mg/l l-蛋氨酸的最小必需培养基中,在30℃下培养4天后,确认表现出蛋氨酸缺陷型和gh1-2基因被pyrf基因取代,得到f09δgh1-2δsc菌株。
[0434]
(4-3)t.cellulolyticus f09δgh1-2δpyrf菌株的制备
[0435]
将t.cellulolyticus f09δgh1-2δsc菌株作为亲本菌株,将gh1-2基因区域插入的pyrf基因用sc基因取代,从而制备f09δgh1-2δpyrf菌株。
[0436]
将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.2和28)的pcr而使gh1-2基因的上游区域扩增,通过使用了引物(seq id no.29和5)的pcr而使gh1-2基因的下游区域扩增。此外,将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.30和31)的pcr而使sc基因的整个区域(包含启动子和终止子)扩增。使用wizard sv gel and pcr clean-up system(promega)而分别使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了crea破坏用dna片段的puc-dgh-sc质粒。将puc-dgh-sc质粒作为模板,通过使用了引物(seq id no.2和5)的pcr而使gh1-2破坏用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0437]
接下来,将f09δgh1-2δsc菌株接种至包含12g/l potato dextrose broth(difco)、20g/l bacto agar(difco)的培养基(以下,称为pd培养基)中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的pyrf破坏用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖、1g/l l尿苷、1g/l尿嘧啶(uracil)的最小必需培养基中,在30℃下培养7天,从而选拔互补了蛋氨酸缺陷型的菌株。将出现的菌落接种至包含1g/l l尿苷、1g/l尿嘧啶(uracil)的最小必需培养基中,在30℃下培养4天后,确认在表现出尿嘧啶(uracil)缺陷
型的同时互补了蛋氨酸缺陷型、和gh1-2基因区域中插入的pyrf基因被sc基因取代,得到f09δgh1-2δpyrf菌株。
[0438]
(4-4)t.cellulolyticus f09δcrea菌株和f09δgh1-2δcrea菌株的制备
[0439]
将t.cellulolyticus f09菌株和f09δgh1-2δpyrf菌株作为亲本菌株,通过以下步骤,破坏crea基因,制备f09δcrea菌株和f09δgh1-2δcrea菌株。
[0440]
crea基因是编码参与分解代谢物阻遏的转录因子的基因。已知crea基因在丝状菌中,参与纤维素酶的表达(mol gen genet.1996jun 24;251(4):451-60,biosci biotechnol biochem.1998dec;62(12):2364-70)。通过crea基因的破坏,而能够使t.cellulolyticus的纤维素酶生产能力提高(wo2015/093467)。
[0441]
首先,根据以下的步骤而制成具有按照t.cellulolyticus的crea基因上游区域、pyrf基因标记、crea基因下游区域的顺序连接的碱基序列的crea破坏用dna片段。
[0442]
将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.32和33)的pcr而使crea基因的上游区域扩增,通过使用了引物(seq id no.34和35)的pcr而使crea基因的下游区域扩增。此外,将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.36和37)的pcr而使pyrf基因的整个区域(包含启动子和终止子)扩增。使用wizard sv gel and pcr clean-up system(promega)而分别使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了crea破坏用dna片段的puc-crea::pyrf质粒。将puc-crea::pyrf质粒作为模板,通过使用了引物(seq id no.32和35)的pcr而使crea破坏用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0443]
接下来,分别将f09菌株和f09δgh1-2δpyrf菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的crea破坏用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖的最小必需培养基中,在30℃下培养7天,从而选拔互补了尿嘧啶(uracil)缺陷型的菌株。将出现的菌落接种至最小必需培养基中,在30℃下培养4天后,确认crea基因被pyrf基因取代,得到f09δcrea菌株和f09δgh1-2δcrea菌株。
[0444]
(4-5)包含龙胆二糖的糖液的制备
[0445]
使用纯化的gh1-2和表达gh1-2的大肠杆菌(e.coli)菌体,制备包含龙胆二糖的糖液。步骤如下所示。
[0446]
在560g/l葡萄糖溶液中,添加实施例(2-1)中得到的纯化gh1-2并使其变为1.4g/l,作为反应液。以使得反应液变得均匀的方式进行搅拌后,在室温下静置52小时。然后,将反应液在95℃下加热10min,作为包含龙胆二糖的糖液。通过所述的离子交换色谱法确认,糖液中包含35.04g/l龙胆二糖。
[0447]
此外,将大肠杆菌(e.coli)ecghh6菌株接种至试验管内的3ml的包含50mg/l卡那霉素的lb培养基中,在37℃、120rpm的条件下进行一晚振荡培养。将该菌体以1/100的量接种至50ml的圆底烧瓶内的包含50mg/l卡那霉素的lb培养基中,在37℃、120rpm的条件下进行振荡培养,直至od600变为0.8。接下来,将圆底烧瓶转移至13℃的箱式振荡器内,在13℃、120rpm的条件下进行1小时振荡培养。然后,添加iptg使其变为1mm,进一步培养17小时。培养后,在冷却至4℃的离心机内在5000rpm下进行5min离心而回收菌体,使其重新悬浮在5ml的培养上清液中。将该5ml的菌体悬浮液投入700g/l葡萄糖溶液100ml中,作为菌体反应液。在以使得菌体反应液变得均匀的方式进行搅拌的同时,使其在室温下进行144小时反应,连续性地取样。在该过程中,随时间经过的同时菌体发生溶菌,从而菌体反应液变为透明(图9左)。就取样的糖液而言,在95℃下加热10min,稀释1000倍而通过所述的离子交换色谱法进行分析。其结果,确认龙胆二糖连续性地生成(图8)。将144小时的反应后的糖液在95℃下加热10min,添加蒸馏水,使其相当于560g/l葡萄糖溶液,作为包含龙胆二糖的糖液。
[0448]
此外,对将以同样方式制备的菌体反应液不搅拌而静置在室温下的情况,以同样的步骤进行评价。其结果,即使在不搅拌的情况下,随时间经过的同时菌体发生溶菌,从而菌体反应液变为透明(图9左)。
[0449]
对于各菌体反应液,将通过0.22μm孔径的过滤器而滤过的那些和未滤过的那些进行sds-page,使用bio-safe
tm coomassie stain(bio-rad)而进行cbb染色,使用pierce fast western blot kit(thermo fisher scientific)和his标签抗体anti-his-tag mab-hrp-direct(mlb)进行western分析。其结果显示,细胞内表达的在c末端添加有his标签的gh1-2漏出至菌体悬浮液中(图9右上)。此外,将通过0.22um孔径的过滤器而滤过的各菌体反应液中用离子交换水进行稀释,通过所述的离子交换色谱法而进行分析,对龙胆二糖进行定量。其结果未发现,因有无搅拌而造成的生成的龙胆二糖的量的差异(图9右下)。
[0450]
(4-6)使用了龙胆二糖的纤维素酶生产培养
[0451]
分别将t.cellulolyticus f09δcrea菌株和f09δgh1-2δcrea菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含20ml的20g/l葡萄糖、24g/l kh2po4、5g/l (nh4)2so4、2g/l尿素、1.2g/lmgso4·
7h2o、0.01g/l znso4·
7h2o、0.01g/l mnso4·
5h2o、0.01g/l cuso4·
5h2o、1g/l玉米浆(c4648,sigma)、1g/l tween 80的液体培养基中,在30℃、220rpm的条件下通过5天的旋转培养而进行预培养。接下来,将15ml的预培养液接种至广口瓶发酵罐中的300ml的包含15g/l葡萄糖、12g/l kh2po4、10g/l(nh4)2so4、1.2g/lmgso4·
7h2o、0.01g/l znso4·
7h2o、0.01g/l mnso4·
5h2o、0.01g/l cuso4·
5h2o、5g/l玉米浆、1g/l tween 80、0.5ml/l disfoam gd(nof)的液体培养基中,将培养温度设为30℃,将通气量设为1/2vvm,通过搅拌而将溶存氧浓度控制为饱和浓度的5%以上,使用氨气将培养ph控制为5,同时进行72小时的流加培养。流加流加液以从培养开始22小时后连续地使培养基中的葡萄糖浓度保持在5~10g/l的范围内。在培养开始72小时后,对培养液进行取样,进行离心分离(15000rpm,5分钟)而得到上
清液。将得到的上清液作为酶液。
[0452]
流加液有8种,其中2种如上所述是包含使用纯化的gh1-2而制备的相当于560g/l葡萄糖的龙胆二糖的糖液(以下,也称为“+gh1-2纯化酶”)和包含使用大肠杆菌(e.coli)ecghh6菌株的菌体而制备的相当于560g/l葡萄糖的加水的龙胆二糖的糖液(以下,也称为“+gh1-2表达大肠杆菌(e.coli)菌体”)。剩余的6种流加液的组成示于表4。
[0453]
[表4]
[0454]
表4
[0455][0456][0457]
通过以下的步骤而测定得到的酶液的滤纸分解活性(fpu/ml),从得到的各酶液在培养中消耗的糖的量计算每碳源的活性收率。
[0458]
<滤纸分解活性(fpu/ml)>
[0459]
在包含裁切成6mm
×
10mm的滤纸(whatman no.1,ge healthcare)的100μl的柠檬酸缓冲液(50mm,ph 5.0)中,添加50μl的适宜稀释的样品,在50℃下进行1小时反应。需要说明的是,不准备进行反应的样品,作为空白组。接下来,添加300μl的dns溶液(1%二硝基水杨酸、20%酒石酸钠
·
钾、0.05%亚硫酸钠、1%氢氧化钠),在95℃下进行5分钟反应后,在冰上冷却5分钟。使反应后的溶液混合,进行离心(12000rpm,5分钟),回收100μl的上清液。然后,测定上清液的540nm的吸光度,减去空白组的值,算出吸光度的增加量。接下来,使用由阶段性地稀释的葡萄糖浓度与540nm的吸光度制成的校准曲线,算出以葡萄糖计在反应溶液中生成的还原糖量。对不同的稀释率的样品进行同样的操作,制成稀释率与葡萄糖生成量的校准曲线,求得生成相当于0.2mg的葡萄糖的还原糖所需要的样品的稀释率,算出稀释前的样品的滤纸分解活性(fpu/ml)。就活性单元而言,将每分钟生成相当于1μmol的葡萄糖的还原糖的酶活性设为“1u”。
[0460]
其结果显示,通过使用龙胆二糖作为流加液中的葡萄糖以外的碳源,而能够与使用纤维二糖的情况相比,实现更高效的纤维素酶生产(图10)。
[0461]
此外,显示,当破坏gh1-2基因时,通过包含龙胆二糖的流加液的添加而能够提高纤维素酶生产效率(图11)。
[0462]
此外,显示,使用纯化的gh1-2而制备的包含相当于560g/l葡萄糖的龙胆二糖的糖液和使用大肠杆菌(e.coli)ecghh6菌株的菌体而制备的包含相当于560g/l葡萄糖的龙胆二糖的糖液,能够实现与使用包含80g/l的纤维二糖的流加液的情况匹敌的高效的纤维素
酶生产(图12)。
[0463]
(5)细胞外分泌型的β-glucosidase bgl3a引起的龙胆二糖的生成
[0464]
上面已经描述了使用定位于细胞内的作为β-glucosidase的gh1-2而制备龙胆二糖的方法。以下表示,使用分泌在细胞外的gh1-2和作为另一主要的β-glucosidase的bgl3a(seq id no.38)而制备龙胆二糖的方法。
[0465]
(5-1)bgl3a基因破坏菌株的制备
[0466]
将t.cellulolyticus f09菌株(日本特开2016-131533)作为亲本菌株,通过以下步骤,破坏编码bgl3a的bgl3a基因,制备f09δbgl3a菌株。
[0467]
首先,根据以下的步骤而制成具有按照t.cellulolyticus的bgl3a基因上游区域、pyrf基因标记、bgl3a基因下游区域的顺序连接的碱基序列的bgl3a破坏用dna片段。
[0468]
将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.39和40)的pcr而使bgl3a基因的上游区域扩增,通过使用了引物(seq id no.41和42)的pcr而使bgl3a基因的下游区域扩增。此外,将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.43和44)的pcr而使pyrf基因的整个区域(包含启动子和终止子)扩增。使用wizard sv gel and pcr clean-up system(promega)而分别使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了bgl3a破坏用dna片段的puc-bgl3a::pyrf质粒。将puc-bgl3a::pyrf质粒作为模板,通过使用了引物(seq id no.45和46)的pcr而使bgl3a破坏用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0469]
接下来,将f09菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的bgl3a破坏用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖的最小必需培养基中,在30℃下培养7天,从而选拔互补了尿嘧啶(uracil)缺陷型的菌株。将出现的菌落接种至最小必需培养基中,在30℃下培养4天后,确认bgl3a基因被pyrf基因取代,得到f09δbgl3a菌株。
[0470]
(5-2)酶液的制备
[0471]
通过将作为纤维素基质的solka-floc(international fiber公司)设为碳源的烧瓶培养而制备t.cellulolyticus f09δbgl3a菌株和f09pyr+菌株生产的纤维素酶酶液。
[0472]
<烧瓶培养>
[0473]
分别将f09δbgl3a菌株和f09pyr+菌株接种至1/2pda平板中,在30℃下培养3天。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘,接种至sf烧瓶培养基(表2)中,在30℃、220rpm的条件下进行7天旋转培养。进行适宜取样,通过0.22μm的针头过滤器而除去菌体,将得到的培养上清液作为酶液。由此,制备源自不包含bgl3a的f09δbgl3a菌株的酶液(以下,也称为
“-
bgl3a”)和源自包含bgl3a的f09pyr+菌株的酶液(以下,也称为“+bgl3a”)。
[0474]
(5-3)从纤维素基质的龙胆二糖的生成
[0475]
使用实施例(5-2)中得到的2种酶液,在包含一定程度的浓度的葡萄糖的条件下,使作为纤维素基质的solka-floc(international fiber公司)分解,分析生成物。就反应而言,将10g的反应液(表5)投入50ml管中,在50℃下以90rpm的速度振荡管的同时进行反应24小时。
[0476]
[表5]
[0477][0478]
结果示于图13。在使用源自不包含bgl3a的f09δbgl3a菌株的酶液的情况下,几乎看不到龙胆二糖的生成,观察到纤维二糖的积蓄。另一方面,在使用源自包含bgl3a的f09pyr+菌株的酶液的情况下,纤维二糖的积蓄降低,确认葡萄糖积蓄的增加和龙胆二糖的生成。此外显示,即使在不添加纤维素基质的条件下,也确认了包含葡萄糖的条件下龙胆二糖的生成,由此出发,bgl3a能够浓度依赖性地从葡萄糖生成龙胆二糖。因此显示,使用包含gh1-2和作为另一主要的β-glucosidase的bgl3a酶的酶液,能够制备龙胆二糖。
[0479]
(5-4)从纤维二糖的龙胆二糖的生成
[0480]
同样,使用作为纤维素基质的分解产物的纤维二糖,进行使用了bgl3a的龙胆二糖生成的研究。
[0481]
使用实施例(5-2)中得到的2种酶液,在包含一定程度的浓度的葡萄糖的条件下使作为纤维素基质的分解产物的纤维二糖(cellobiose)分解,分析生成物。就反应而言,将10g的反应液(表6)投入50ml管中,在50℃下以90rpm的速度振荡管的同时进行反应24小时。
[0482]
[表61
[0483][0484]
结果示于图14。在使用源自包含bgl3a的f09pyr+菌株的酶液的情况下,能够确认从作为纤维二糖基质的分解产物的纤维二糖出发的龙胆二糖的生成。另一方面,在使用源自不包含bgl3a的f09δbgl3a菌株的酶液的情况下,不仅纤维二糖的分解降低,而且没有确认到龙胆二糖的生成。该结果显示,bgl3a从纤维二糖生成龙胆二糖。此外,与实施例(5-3)的结果结合,显示通过使用包含bgl3a的t.cellulolyticus的菌体培养液,而能够从纤维素基质借助纤维二糖而生成龙胆二糖。
[0485]
(6)gh1-2活性弱化菌株带来的纤维素酶生产培养评价
[0486]
显示在实施例(4-6)中,通过破坏gh1-2基因而能够提高龙胆二糖的添加带来的纤维素酶生产效率。因此,接下来,研究是否能够通过使gh1-2基因产物(gh1-2)的活性降低,而提高各种条件中的纤维素酶生产效率。
[0487]
(6-1)变异型gh1-2的制备
[0488]
将实施例(2-1)中制备的pet24a-gh1-2-his6作为模板,使用wizard sv gel and pcr clean-up system(promega)对通过使用了引物(seq id no.52和53)的pcr而扩增的pcr产物进行纯化。通过in-fusion hd cloning kit(takara bio)而连接纯化的pcr产物。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含50mg/l卡那霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了具有gh1-2的363位色氨酸被苯丙氨酸取代的w363f变异的变异型gh1-2基因的cdna片段的pet24a-gh1-2(w363f)-his6质粒。此外,使用引物(seq id no.54和55),通过同样的步骤,而得到导入了具有gh1-2的449位的色氨酸被苯丙氨酸取代的w449f变异的变异型gh1-2基因的cdna片段的pet24a-gh1-2(w449f)-his6质粒。
[0489]
分别将pet24a-gh1-2(w363f)-his6质粒和pet24a-gh1-2(w449f)-his6质粒转化为大肠杆菌(e.coli)rosetta
tm 2(de3)plyss competent cells(novagen),在lb琼脂培养基(包含50mg/l卡那霉素)中,在37℃下培养一晚,从而形成菌落。将形成的菌落在lb琼脂培养基(包含50mg/l卡那霉素)上划线,取得单个菌落,从而得到ecghw363fh6菌株和ecghw449fh6菌株。
[0490]
分别将ecghw363fh6菌株和ecghw449fh6菌株接种至试验管内的3ml的包含50mg/l卡那霉素的lb培养基中,在37℃、120rpm的条件下进行一晚振荡培养。将培养液以1/100量接种至50ml的圆底烧瓶内的包含50mg/l卡那霉素的lb培养基中,在37℃、120rpm的条件下进行振荡培养,直至od600变为0.8。接下来,将圆底烧瓶转移至13℃的箱式振荡器内,在13℃、120rpm的条件下进行1小时振荡培养。然后,添加iptg使其变为1mm,进一步培养17小时。培养后,在4℃、5000rpm的条件下进行5min离心而回收菌体,在-20℃的冷柜中冻结1小时。然后,使用ni-nta fast start kit(qiagen)而在非改性条件下从菌体中萃取可溶性蛋白质,使用试剂盒中随附的ni-nta亲和柱、清洗缓冲液和溶出缓冲液而使c末端添加有his标
签的2种变异型gh1-2分别纯化。包含纯化gh1-2的溶出部分使用amicon ultra-15 30kda cut off(merck millipore)超滤膜过滤器和50mm磷酸钠缓冲液(ph=6.5)而进行脱盐和浓缩。确认通过sds-page而纯化的2种变异型gh1-2是单条带,用于之后的实验。
[0491]
(6-2)变异型gh1-2的活性评价
[0492]
对纯化的2种变异型gh1-2和实施例(2-1)中得到的纯化的野生型gh1-2的水解活性进行比较。首先,使野生型gh1-2和变异型gh1-2的酶溶液的蛋白质浓度一致。并且,稀释酶溶液并使得反应后样品的波长410nm中的吸光度与校准曲线匹配,得到稀释酶液。将稀释酶液10μl添加至1ml的基质液(含有10mm的p-nitrophenyl-β-d-glucopyranoside(wako pure chemical industry)的50mm磷酸钠缓冲液(ph 6.5))中,在45℃下培养(incubate)10分钟。然后在95℃下进行10分钟培养(incubate),从而使酶失活,冷却至室温之后,测定在波长410nm下的吸光度。作为背景而减去未进行培养(incubate)而仅在95℃下进行10分钟的失活处理的样品的测定结果。就校准曲线而言,通过用50mm的磷酸钠缓冲液稀释p-nitrophenol(fluka chemical公司)并测定吸光度而制成。将p-nitrophenol的生成量作为水解活性的指标,将野生型gh1-2的水解活性设为100%作为相对值而算出各gh1-2的水解活性。其结果显示,2种变异型gh1-2(w363f和w449f)均水解活性降低(图15)。
[0493]
此外,将纯化的2种变异型gh1-2与实施例(2-1)中得到的纯化的野生型gh1-2的纤维二糖(cellobiose)反应时的水解物与糖转移生成物进行比较。在实施例(3-2)所述的方法中,通过tlc而使反应液展开和显色。结果示于图16。显示2种变异型gh1-2(w363f和w449f)均对纤维二糖(cellobiose)的水解活性降低。在使用更多(5倍)w363f变异型gh1-2的情况下,观察到与使用野生型gh1-2的情况接近的展开的图案,暗示糖转移生成物的生成也同样降低。在使用w449f变异型gh1-2的情况下,几乎没有观察到纤维二糖(cellobiose)的水解和糖转移,观察到非常轻微的纤维二糖(cellobiose)水解而生成葡萄糖。
[0494]
在实施例(2-2)所述的条件下将使用野生型gh1-2时的所述的反应液供于离子交换色谱法,分析反应生成物,从而观察纤维二糖(cellobiose)的水解物和糖转移生成物随时间经过的变化。其结果显示,在野生型gh1-2的情况下,在龙胆二糖之前先生成纤维三糖、纤维四糖,并且在龙胆二糖之前,纤维二糖(cellobiose)等的纤维寡糖先被分解(图17)。
[0495]
同样,在实施例(2-2)所述的条件下将使用w363f变异型gh1-2时的所述的反应液供于离子交换色谱法,分析反应生成物,从而观察纤维二糖(cellobiose)的水解物和糖转移生成物随时间经过的变化,与使用野生型gh1-2的情况比较。其结果显示,在w363f变异型gh1-2的情况下,纤维二糖(cellobiose)的水解效率降低,除此之外,纤维三糖、龙胆二糖的生成延迟(图18)。即,显示在w363f变异型gh1-2的情况下,不仅对纤维二糖(cellobiose)的水解效率降低,而且糖转移生成物的生成效率也降低。
[0496]
认为在具有这样的变异型gh1-2的菌株中,组入细胞内的纤维二糖(cellobiose)、其糖转移生成物不易水解成葡萄糖,因此,使细胞质中的存在量增大。即,在具有这样的变异型gh1-2的菌株中,稳定性地供给诱导纤维素酶基因的表达的龙胆二糖等糖转移生成物,因此推测纤维素酶的表达提高,另一方面,在具有野生型gh1-2的菌株中,在细胞质中从纤维二糖(cellobiose)生成糖转移生成物的效率较高,另一方面,作为其基质的纤维二糖(cellobiose)、糖转移生成物水解的效率也显著较高,因此推测诱导纤维素酶基因的表达的龙胆二糖等糖转移生成物的供给变得不稳定,因此,纤维素酶的表达受到制限。
[0497]
(6-3)t.cellulolyticus的过去的培育中的gh1-2基因内的有效变异c267p的发现
[0498]
将t.cellulolyticus y-94菌株(ferm bp-5826)、tn菌株(ferm bp-685)和f09菌株的基因组dna作为模板,使用引物(seq id no.16、17、18、19、20、21)而进行gh1-2基因区域的序列分析。其结果可知,在从y-94菌株构建tn菌株的培育的过程中,将gh1-2的267位的半胱氨酸被脯氨酸取代的c267p变异导入gh1-2基因中(图19)。在日本特开2011-193773中报告了,tn菌株存在2513个的变异候补位点。然而,在日本特开2011-193773没有提及该c267p变异。
[0499]
(6-4)c267p变异向t.cellulolyticus y-94菌株的gh1-2基因的导入
[0500]
将t.cellulolyticus的tn菌株(ferm bp-685)的基因组dna作为模板,通过使用了引物(seq id no.56和57)的pcr而使包含c267p变异的gh1-2基因的从上游4kb至下游4kb的dna片段扩增。使用wizard sv gel andpcr clean-up system(promega)使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了所述dna片段的puc-gh1-2tn质粒。
[0501]
接下来,将puc-gh1-2tn质粒作为模板,通过使用了引物(seq id no.58和59)的pcr而使puc-gh1-2tn质粒在gh1-2基因的终始密码子的下游约0.5kb位点开裂的片段扩增。此外,将pcdna3.1/hygro(+)质粒(invitrogen)作为模板,通过使用了引物(seq id no.60和61)的pcr而使潮霉素抗性基因盒区域(包含启动子和终止子)扩增。使用wizard sv gel and pcr clean-up system(promega)而分别使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了包含c267p变异的gh1-2基因的从上游约4kb至下游约4kb区域和潮霉素抗性基因的puc-c267prep质粒。将puc-c267prep质粒作为模板,通过使用了引物(seq id no.56和57)的pcr而使用于取代gh1-2基因区域的dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0502]
接下来,将y-94菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的用于取代gh1-2基因区域的dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种在包含1m蔗糖的最小必需培养基(表1)中,在30℃下培养1天后,覆盖包含0.5g/l hygromycin b和1m蔗糖的最小必需培养基(表1),在30℃下培养7天,从而选拔获得潮霉素抗性的菌株。将出
现的菌落接种至包含0.5g/l hygromycin b的最小必需培养基中,在30℃下培养4天后,确认gh1-2基因区域被包含c267p变异的gh1-2基因区域取代,得到c267prep菌株。
[0503]
(6-5)基于烧瓶培养的c267prep菌株的纤维素酶生产评价
[0504]
根据实施例(1-2)所述的步骤,通过将作为纤维素基质的solka-floc(international fiber公司)设为碳源的烧瓶培养而对基于y-94菌株、c267prep菌株和tn菌株的纤维素酶生产进行评价。将培养上清液中的总蛋白质浓度作为纤维素酶生产量的指标。其结果显示,与作为亲本菌株的y-94菌株相比,c267prep菌株和tn菌株的纤维素酶的分泌生产能力和纤维素的同化能力显著上升(图20)。
[0505]
(6-6)基于晕圈测定的c267prep菌株的纤维素酶生产评价
[0506]
根据实施例(1-4)所述的步骤,通过将作为纤维素基质的cmc设为碳源的晕圈测定而对基于y-94菌株、c267prep菌株和tn菌株的纤维素酶生产进行评价。其结果显示,与作为亲本菌株的y-94菌株相比,c267prep菌株和tn菌株的纤维素酶的分泌生产能力和纤维素的同化能力显著上升(图21)。
[0507]
(6-7)w363f变异和w449f变异向t.cellulolyticus y-94菌株的gh1-2基因的导入
[0508]
将t.cellulolyticus y-94菌株(ferm bp-685)的基因组dna作为模板,通过使用了引物(seq id no.56和57)的pcr而使gh1-2基因的从上游4kb至下游4kb的dna片段扩增。使用wizard sv gel and pcr clean-up system(promega)使pcr产物纯化。通过in-fusion hd cloning kit(takarabio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了所述dna片段的puc-gh1-2wt质粒。
[0509]
接下来,将puc-gh1-2wt质粒作为模板,通过使用了引物(seq id no.58和59)的pcr而使puc-gh1-2wt质粒在gh1-2基因的终始密码子的下游约0.5kb位点开裂的片段扩增。此外,将pcdna3.1/hygro(+)质粒(invitrogen)作为模板,通过使用了引物(seq id no.60和61)的pcr而使潮霉素抗性基因盒区域(包含启动子和终止子)扩增。使用wizard sv gel and pcr clean-up system(promega)而分别使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了gh1-2基因的从上游约4kb至下游约4kb区域和潮霉素抗性基因的puc-wtrep质粒。
[0510]
此外,将puc-wtrep质粒作为模板,使用wizard sv gel and pcr clean-up system(promega)使通过使用了引物(seq id no.52和53)的pcr而扩增的pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而连接纯化的pcr产物。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含50mg/l卡那霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了包含w363f变异的gh1-2基因的从上游约4kb至下游约4kb区域和潮霉素抗性基因的puc-w363frep质粒。此外,使用引物(seq id no.54和55)而通过同样的步骤,得到导入了包含w449f变异的gh1-2基因的从上游约4kb至下游约4kb区域和潮霉素抗性基因的puc-w449frep质粒。
[0511]
分别将puc-w363frep质粒和puc-w449frep质粒作为模板,通过使用了引物(seq id no.56和57)的pcr而使用于取代gh1-2基因区域的dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0512]
接下来,将y-94菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的用于取代gh1-2基因区域的dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种在包含1m蔗糖的最小必需培养基(表1)中,在30℃下培养1天后,覆盖包含0.5g/l hygromycin b和1m蔗糖的最小必需培养基(表1),在30℃下培养7天,从而选拔获得潮霉素抗性的菌株。将出现的菌落接种至包含0.5g/l hygromycin b的最小必需培养基中,在30℃下培养4天后,确认gh1-2基因区域被包含w363f变异或w449f变异的gh1-2基因区域取代,得到w363frep菌株和w449frep菌株。
[0513]
(6-8)基于烧瓶培养的w363frep菌株和w449frep菌株的纤维素酶生产评价
[0514]
根据实施例(1-2)所述的步骤,通过将作为纤维素基质的solka-floc(international fiber公司)设为碳源的烧瓶培养而对基于y-94菌株、c267prep菌株、w363frep菌株和w449frep菌株的纤维素酶生产进行评价。将培养上清液中的总蛋白质浓度和纤维素酶比活性作为纤维素酶生产量的指标。其结果显示,与作为亲本菌株的y-94菌株相比,w363frep菌株和w449frep菌株的纤维素酶的分泌生产能力和纤维素的同化能力显著上升(图22)。
[0515]
(6-9)基于晕圈测定的w363frep菌株、w449frep菌株和c267prep菌株的纤维素酶生产评价
[0516]
根据实施例(1-4)所述的步骤,分别使y-94菌株、w363frep菌株、w449frep菌株和c267prep菌株在将cmc设为碳源的最小必需培养基上和将包含1mm的龙胆二糖的cmc设为碳源的最小必需培养基上,生长5天。培养后,添加以10:1混合50mm磷酸钠缓冲液(ph 7.0)与2mg/ml的congo red(nacalai tesque)而得到的congo red溶液,使其涂覆平板表面,在室温下静置30分钟。然后,除去平板上的congo red溶液,添加磷酸钠缓冲液(50mm,ph 7.0),使其涂覆平板表面。再次,在室温下静置30分钟后,除去平板上的磷酸钠缓冲液,稍稍使平板表面干燥,测定可视化的晕圈的直径与宽度。
[0517]
结果示于图23。再次显示,与作为亲本菌株的y-94菌株相比,就w363frep菌株、w449frep菌株和c267prep菌株而言,在不添加龙胆二糖的情况和添加的情况中的任一种情况都使分解晕圈的直径和宽度显著扩大,纤维素酶的分泌生产能力和纤维素的同化能力显著上升。此外,显示,与作为亲本菌株的y-94菌株相比,就w363frep菌株、w449frep菌株和
c267prep菌株而言,基于龙胆二糖的添加的分解晕圈的直径和宽度扩大的程度增大。该结果显示,通过龙胆二糖的添加与gh1-2基因中的这些变异的组合而使得纤维素酶的分泌生产相乘性地提高。
[0518]
(7)gh1-2和bgl3a双重破坏菌株中的使用龙胆二糖的培养
[0519]
(7-1)f09δghδbgl3a菌株的制备
[0520]
将实施例(4-3)中制备的t.cellulolyticus f09δgh1-2δpyrf菌株作为亲本菌株,通过以下步骤,破坏编码bgl3a的bgl3a基因,制备f09δgh1-2δbgl3a菌株。
[0521]
首先,将实施例(5-1)中制备的puc-bgl3a::pyrf质粒作为模板,通过使用了引物(seq id no.45和46)的pcr而使bgl3a破坏用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0522]
接下来,将f09δgh1-2δpyrf菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalase(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的bgl3a破坏用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖的最小必需培养基中,在30℃下培养7天,从而选拔互补了尿嘧啶(uracil)缺陷型的菌株。将出现的菌落接种至最小必需培养基中,在30℃下培养4天后,确认bgl3a基因被pyrf基因取代,得到f09δgh1-2δbgl3a菌株。
[0523]
(7-2)gh1-2和bgl3a双重破坏菌株的纤维素酶生产平板晕圈测定评价
[0524]
根据实施例(1-4)所述的步骤,分别使实施例(1-1)中制备的f09pyrf+菌株和f09δgh1-2菌株、实施例(5-1)中制备的f09δbgl3a菌株、和实施例(7-1)中制备的f09δgh1-2δbgl3a菌株在将cmc设为碳源的最小必需培养基上和将包含1mm的龙胆二糖的cmc设为碳源的最小必需培养基上生长5天。培养后,添加以10:1混合50mm磷酸钠缓冲液(ph 7.0)与2mg/ml的congo red(nacalai tesque)而得到的congo red溶液,使其涂覆平板表面,在室温下静置30分钟。然后,除去平板上的congo red溶液,添加磷酸钠缓冲液(50mm,ph 7.0),使其涂覆平板表面。再次,在室温下静置30分钟后,除去平板上的磷酸钠缓冲液,稍稍使平板表面干燥,测定可视化的晕圈的直径和宽度。
[0525]
结果示于图24。显示与f09pyrf+菌株、f09δgh1-2菌株和f09δbgl3a菌株相比,就作为双重破坏菌株的f09δgh1-2δbgl3a菌株而言,在添加龙胆二糖的情况下的分解晕圈的宽度扩大,纤维素酶的分泌生产能力上升。该结果显示,通过龙胆二糖的添加和gh1-2基因与bgl3a基因的双重破坏的组合而使得纤维素酶的分泌生产相乘性地提高。即,暗示,通过龙胆二糖等的表达诱导物质的添加和gh1-2的活性降低、和bgl3a等的其他β-glucosidase的活性降低的组合,而使得纤维素酶的分泌生产相乘性地提高。
[0526]
(8)异源蛋白质的表达
[0527]
(8-1)talaromyces cellulolyticus的yscb基因损失菌株f09δyscb的构建
[0528]
将talaromyces cellulolyticus f09菌株(日本特开2016-131533)作为亲本菌株,通过以下步骤而破坏yscb基因(seq id no.62),构建t.cellulolyticus f09δyscb菌株。
[0529]
首先,根据以下的步骤而制成具有按照t.cellulolyticus的yscb基因的上游区域、潮霉素抗性基因、t.cellulolyticus的yscb基因下游区域的顺序连接的碱基序列的yscb基因破坏用dna片段。
[0530]
将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了引物(seq id no.63和64)的pcr而使yscb基因的上游区域扩增,通过使用了引物(seq id no.65和66)的pcr而使yscb基因的下游区域扩增。此外,将搭载有潮霉素抗性基因的pcdna3.1/hygro(+)(life technologies)作为模板,通过使用了引物(seq id no.67和68)的pcr而使潮霉素抗性基因(包含启动子和终止子)扩增。使用wizard sv gel and pcr clean-up system(promega)使pcr产物纯化。通过in-fusion hd cloning kit(takara bio)而将纯化的pcr产物导入并与试剂盒随附的puc质粒连接。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了yscb基因破坏用dna片段的puc-yscb::hyg质粒。将puc-yscb::hyg质粒作为模板,通过使用了引物(seq id no.63和66)的pcr而使yscb基因破坏用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。
[0531]
接下来,将f09菌株接种至包含12g/l potato dextrose broth(difco)、20g/l bacto agar(difco)的培养基中,在30℃下进行培养。将用吸管在琼脂培养基上形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含24g/l potato dextrose broth的培养基中,在30℃、220rpm的条件下进行2天旋转培养。通过离心分离(5000rpm,5分钟)而回收菌体,添加包含10g/l yatalas e(takara bio)、10mm kh2po4、0.8m nacl的水溶液(ph6.0)30ml,在振荡的同时使其在30℃下进行2小时反应,消化细胞壁而进行原生质体化。通过玻璃过滤器而除去残渣后,通过离心分离(2000rpm,10分钟)而回收原生质体,通过包含1.2m sorbitol、10mm cacl2的tris-hcl缓冲液(ph7.5)而悬浮成1ml,制备原生质体溶液。在200μl的原生质体溶液中,添加10μg纯化的yscb破坏用dna片段和50μl包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5),在冰上放置30分钟。然后进一步添加1ml的包含400g/l peg4000、10mm cacl2的tris-hcl缓冲液(ph7.5)并进行混合,在室温下放置15分钟而进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖、1g/l尿嘧啶(uracil)、1g/l尿苷的最小必需培养基(10g/l葡萄糖、10mm nh4cl、10mm kh2po4、7mm kcl、2mmmgso4、0.06mg/l h3bo3、0.26mg/l (nh4)6mo7o
24
·
4h2o、1mg/l fecl3·
6h2o、0.4mg/l cuso4·
5h2o、0.08mg/l mncl2、2mg/l zncl2、20g/l bacto agar)中,在30℃下培养1天后,覆盖包含0.5g/l hygromycinb、24g/l potato dextrose broth、7g/l bacto agar的培养基,进一步在30℃下培养3天,从而选拔潮霉素抗性菌株。将出现的菌落接种至包含0.5g/l hygromycin b的最小必需培养基中,在30℃下培养4天后,确认yscb基因被hygromycin抗性基因取代,得到源自f09的yscb破坏菌株(f09δyscb菌株)。
[0532]
(8-2)人血清白蛋白(hsa)表达菌株的构建
[0533]
将t.cellulolyticus f09菌株和f09δyscb菌株作为亲本菌株,通过以下步骤而构建人血清白蛋白(hsa)表达菌株。
[0534]
首先,根据以下的步骤而制成具有按照t.cellulolyticus的crea基因上游区域、t.cellulolyticus的cbh2基因上游区域(cbh2启动子;seq id no.50)、t.cellulolyticus的cbh1分泌信号编码序列(seq id no.70)、hsa基因(seqid no.71)、t.cellulolyticus的cbh2基因下游区域(cbh2终止子;seq id no.72)、t.cellulolyticus的pyrf基因标记(seq id no.73)、t.cellulolyticus的crea基因下游区域的顺序连接的碱基序列的hsa表达用dna片段。
[0535]
将t.cellulolyticus y-94菌株(ferm bp-5826)的基因组dna作为模板,通过使用了(seq id no.74和75)的pcr而使crea基因的上游区域扩增,通过使用了(seq id no.76和77)的pcr而使cbh2基因的上游区域扩增,通过使用了(seq id no.78和79)的pcr而使cbh1分泌信号编码序列扩增,通过使用了(seq id no.80和81)的pcr而使cbh2基因的下游区域扩增,通过使用了(seq id no.82和83)的pcr而使pyrf基因标记扩增,通过使用了(seq id no.84和85)的pcr而使crea基因下游区域扩增。此外,将从eurofins株式会社购入的全合成基因作为模板,通过使用了引物(seq id no.86和87)的pcr而使hsa基因扩增。使用wizard sv gel and pcr clean-up system(promega)使pcr产物纯化。将混合2种纯化的pcr产物而得到的混合物作为模板,再次进行pcr并连接,重复后,通过in-fusion hd cloning kit(takara bio)而导入试剂盒随附的puc质粒中。用反应物转化大肠杆菌(e.coli)jm109,在lb琼脂培养基(包含100mg/l氨苄青霉素)中在37℃下培养一晚,从而形成菌落。使用wizard plus miniprep system(promega),由得到的转化体得到导入了hsa表达用dna片段的puc-crea::pcbh2-hsa-pyrf质粒。将puc-crea::pcbh2-hsa-pyrf质粒作为模板,通过使用了引物(seq id no.76和85)的pcr而使hsa表达用dna片段扩增,通过乙醇沉淀而进行浓缩和纯化。需要说明的是,通过使crea基因的上游和下游序列与hsa表达序列的两端连接,可以不是在基因组上的随机的位置,而是针对crea基因区域而插入hsa表达序列。
[0536]
接下来,以与(8-1)同样的手法培养f09菌株和f09δyscb菌株,进行原生质体化,以与(8-1)同样的方式用纯化的hsa表达用dna片段进行转化。将通过离心分离(2000rpm,10分钟)而回收的原生质体播种至包含1m蔗糖的最小必需培养基中,在30℃下培养7天,从而选拔互补了尿嘧啶(uracil)缺陷型的菌株。将出现的菌落接种至最小必需培养基中,在30℃下培养4天后,确认crea基因区域被hsa表达序列取代,得到源自f09菌株和f09δyscb菌株的hsa表达菌株。
[0537]
(8-3)人血清白蛋白(hsa)表达菌株的使用了龙胆二糖的培养
[0538]
为了确认添加龙胆二糖同时进行培养时的hsa的分泌生产,对源自f09δyscb菌株的hsa表达菌株进行流加培养。
[0539]
将源自t.cellulolyticus f09δyscb菌株的hsa表达菌株接种至pd培养基中,在30℃下进行培养。将用吸管在形成的菌落的末端附近打孔而得到的1个琼脂盘接种至包含20ml的20g/l葡萄糖、24g/l kh2po4、5g/l(nh4)2so4、2g/l尿素、1.2g/lmgso4·
7h2o、0.01g/l znso4·
7h2o、0.01g/l mnso4·
5h2o、0.01g/l cuso4·
5h2o、1g/l玉米浆(c4648,sigma)、1g/l tween 80的液体培养基中,在30℃、220rpm的条件下通过5天的旋转培养而进行预培养。
接下来,将15ml的预培养液接种至广口瓶发酵罐中的300ml的包含15g/l葡萄糖、12g/l kh2po4、10g/l(nh4)2so4、1.2g/lmgso4·
7h2o、0.01g/l znso4·
7h2o、0.01g/l mnso4·
5h2o、0.01g/l cu so4·
5h2o、5g/l玉米浆、1g/l tween 80、0.5ml/l disfoam gd(nof)的液体培养基中,将培养温度设为30℃,将通气量设为1/2vvm,通过搅拌而将溶存氧浓度控制为饱和浓度的5%以上,使用氨气将培养ph控制为5,同时进行45小时的流加培养。流加流加液,使得从培养开始22小时后连续地使培养基中的葡萄糖浓度保持在5~10g/l的范围内。在培养开始45小时后,对培养液进行取样,进行离心分离(15000rpm,5分钟)而得到上清液。将得到的上清液作为培养上清液样品溶液。流加液有2种,组成示于表7。
[0540]
[表7]
[0541] 葡萄糖(g/l)龙胆二糖(g/l)纤维二糖(g/l)龙胆二糖80g/l480800纤维二糖80g/l480080
[0542]
使用albumin elisa quantitation kit,human(bethyl laboratories,inc.),进行基于elisa的hsa的定量。其结果确认,与流下纤维二糖(cellobiose)的情况相比,流下龙胆二糖的情况下,hsa分泌生产量增加(图25)。该结果显示,通过龙胆二糖的添加而使基于龙胆二糖诱导性启动子的控制下的异源蛋白质的分泌生产提高。
[0543]
工业实用性
[0544]
通过本发明,而能够高效地制造纤维素酶等的蛋白质。此外,通过本发明,而能够高效地制造龙胆二糖等的二糖。
[0545]
<序列表的说明>
[0546]
seq id no.1:talaromyces cellulolyticus y-94菌株的gh1-2基因的碱基序列
[0547]
seq id no.2~21:引物
[0548]
seq id no.22:talaromyces cellulolyticus y-94菌株的gh1-2基因的cdna的碱基序列
[0549]
seq id no.23:talaromyces cellulolyticus y-94菌株的gh1-2蛋白的氨基酸序列
[0550]
seq id no.24~37:引物
[0551]
seq id no.38:talaromyces cellulolyticus s6-25菌株的bgl3a蛋白的氨基酸序列
[0552]
seq id no.39~46:引物
[0553]
seq id no.47:talaromyces cellulolyticus s6-25菌株的crea基因的碱基序列
[0554]
seq id no.48:talaromyces cellulolyticus s6-25菌株的bgl3a基因的碱基序列
[0555]
seq id no.49:talaromyces cellulolyticus的cbh1启动子的碱基序列
[0556]
seq id no.50:talaromyces cellulolyticus的cbh2启动子的碱基序列
[0557]
seq id no.51:talaromyces cellulolyticus的cbh1信号肽的氨基酸序列
[0558]
seq id no.52~61:引物
[0559]
seq id no.62:talaromyces cellulolyticus s6-25菌株的yscb基因的碱基序列
[0560]
seq id no.63~68:引物
[0561]
seq id no.69:talaromyces cellulolyticus s6-25菌株的yscb蛋白的氨基酸序列
[0562]
seq id no.70:talaromyces cellulolyticus的编码cbh1信号肽的碱基序列
[0563]
seq id no.71:人血清白蛋白(hsa)基因的碱基序列
[0564]
seq id no.72:t.cellulolyticus的cbh2终止子的碱基序列
[0565]
seq id no.73:t.cellulolyticus的pyrf基因标记的碱基序列
[0566]
seq id no.74~87:引物。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
相关标签:

 热门咨询
热门咨询




tips