
 商标分类
商标分类  商标转让
商标转让 
MFI拓扑学结构硅分子筛及其制备方法和应用与流程
 2021-01-30 20:01:44|
2021-01-30 20:01:44| 323|
323| 起点商标网
起点商标网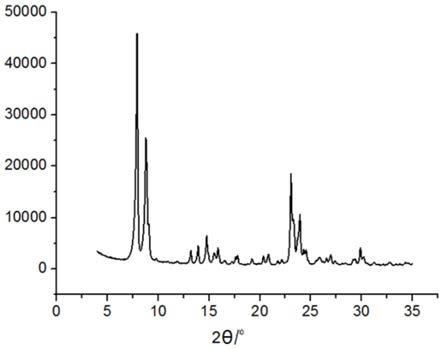
mfi拓扑学结构硅分子筛及其制备方法和应用
技术领域
[0001]
本发明涉及硅分子筛领域,具体涉及mfi拓扑学结构硅分子筛及其制备方法和应用。
背景技术:
[0002]
silicalite-1分子筛又称全硅、纯硅分子筛,于1978年由美国联合碳化物公司的e.m.flanigen等首次被成功合成出来,属于“pentasil”家族的成员之一。全硅分子筛是一种具有mfi拓扑学结构的无铝分子筛,是zsm-5型结构分子筛家族中组成最简单的一种分子筛,其骨架仅含有硅原子和氧原子,基本结构单元为sio
4
四面体。mfi拓扑学结构的全硅分子筛拥有丰富的微孔结构和规整均匀的三维细孔道,具有确定的zsm-5型分子筛的晶体结构,较高的内比表面积,良好的热稳定性、吸附和脱附能力等性能。全硅分子筛在膜吸附分离、净化、催化材料等领域的开发应用正受到人们的日益重视。
[0003]
全硅分子筛可作膜分离的材料,也可作环己酮肟气相贝克曼重排反应生产己内酰胺的催化剂。全硅分子筛的合成方法一般采用传统的有机原料水热法,硅源多选用固体氧化硅、硅溶胶、白炭黑或正硅酸乙酯(简称teos)等,模板剂多选用四丙基氢氧化铵(简称tpaoh)、低碳烃类季铵盐或胺类化合物等,在150℃以上的温度下晶化若干天。现有技术合成的全硅分子筛,无定形氧化硅含量较多,相对结晶度较差且晶体颗粒较大。
[0004]
现有技术中硅/铝比达到50000以上的全硅分子筛通常选择正硅酸乙酯作硅源,四丙基氢氧化铵作模板剂和碱源。
[0005]
美国专利申请us4061724a中公开的全硅分子筛,它的制备原料中不含有铝源,只有硅源、碱源、模板剂和水,不同于抽取骨架铝而形成的全硅分子筛,其是直接合成全硅分子筛,具有mfi拓扑学晶体结构。这种全硅分子筛所用硅源为硅溶胶、硅凝胶或白碳黑中的一种,它是由摩尔组成为150-700h
2
o:13-50sio
2
:0-6.5m
2
o:q
2
o的反应混合物在100~250℃、自生压力下水热晶化50~150小时合成的,其中,m是碱金属,q是分子式为r
4
x
+
的季阳离子,r代表氢或有2-6个碳原子的烷基,x是磷或氮。
[0006]
jp59164617a中公开的mfi结构全硅分子筛,是以正硅酸乙酯为硅源,四丙基氢氧化铵为模板剂和碱源制备的。在catal.rev.-sci.eng.,39(4),395-424(1997)中的研究表明,以正硅酸乙酯为硅源合成的全硅分子筛具有较高的bet总比表面和外表面积,可分别达到400米
2
/克和20~40米
2
/克以上,且环己酮肟的转化率和己内酰胺的选择性与外表面积的增加成正比。
[0007]
cn102050464b公开了一种硅分子筛的合成方法,其特征在于合成过程包括下列步骤:(1)将正硅酸乙酯与四丙基氢氧化铵在室温下混合、搅拌、充分水解,并补加水,形成摩尔组成为tpaoh/sio
2
=0.05~0.5,etoh/sio
2
=4,h
2
o/sio
2
=5~100的混合物;(2)将上述混合物在密闭反应釜中,自生压力下80~120℃晶化0.5~10天,然后过滤、洗涤、干燥,400~600℃焙烧1~10小时,得到硅分子筛。该方法所得到的分子筛用于环己酮肟气相贝克曼重排反应制备己内酰胺时,具有很高的环己酮肟转化率和己内酰胺选择性。
[0008]
cn107335465a公开了一种含贵金属离子的silicate-1分子筛催化剂的制备方法,该方法包括:a、将硅源、贵金属源、有机模板剂和水混合,得到胶体混合物,其中,以摩尔比计,所述胶体混合物中sio
2
:贵金属离子:有机模板剂:h
2
o=1:(2.6
×
10-8-1
×
10-6
):(0.05-0.50):(5-100);b、将步骤a中得到的所述胶体混合物进行水热晶化,得到晶化产物;c、将步骤b中得到的所述晶化产物进行洗涤、分离处理,得到含贵金属离子的silicate-1分子筛;d、将步骤c中得到的所述含贵金属离子的silicate-1分子筛进行成型处理、焙烧处理和含氮化合物的碱性缓冲溶液后处理,再进行洗涤、分离、干燥,得到含贵金属离子的silicate-1分子筛催化剂。
[0009]
虽然现有技术可以成功合成全硅分子筛,将现有的mfi拓扑学结构的全硅分子筛作为催化剂用于环己酮肟气相贝克曼重排反应可以使得环己酮肟转化率和己内酰胺选择性得到一定的提高,但为了提升环己酮肟气相贝克曼重排工艺技术的经济性,有必要开发出一款新的硅分子筛的合成方法。
技术实现要素:
[0010]
本发明的目的是为了提供一种mfi拓扑学结构硅分子筛及其制备方法和应用,将本发明提供的分子筛用于环己酮肟气相贝克曼重排反应过程中,可以提高环己酮肟的转化率和己内酰胺的选择性,提升环己酮肟气相贝克曼重排工艺技术的经济性。
[0011]
本发明第一方面提供一种mfi拓扑学结构硅分子筛,该分子筛含有硅元素、氧元素和金属元素,以分子筛的总量为基准,金属元素的含量为5-100μg/g;该分子筛的bet比表面积为400-500m
2
/g,所述金属元素选自过渡金属元素和iiia族金属元素中的至少一种。
[0012]
本发明第二方面提供一种mfi拓扑学结构硅分子筛的制备方法,该方法包括:
[0013]
(1)将硅源、有机胺、有机模板剂、金属源、有机醇和水混合,得到胶体混合物,其中,硅源、有机胺、有机模板剂、有机醇和水的用量摩尔比为1:(0.05-0.5):(0.05-0.5):(4-20):(5-100),硅源与金属源的用量质量比为(10000-200000):1,硅源以sio
2
计,金属源以金属元素计;
[0014]
(2)将所述胶体混合物进行两段变温醇-水热体系晶化,所述两段变温醇-水热体系晶化的条件包括:在40-70℃下晶化0.5-5天,然后在80-130℃下晶化0.5-5天;
[0015]
(3)将步骤(2)得到的晶化母液进行过滤、干燥和焙烧;
[0016]
所述金属选自过渡金属和iiia族金属中的至少一种。
[0017]
优选地,所述有机胺为三正丙胺。
[0018]
优选地,所述有机模板剂为四丙基氢氧化铵和/或四乙基氢氧化铵。
[0019]
本发明第三方面提供上述制备方法制得的mfi拓扑学结构硅分子筛。
[0020]
本发明第四方面提供上述mfi拓扑学结构硅分子筛在环己酮肟气相贝克曼重排反应中的应用。
[0021]
本发明提供的分子筛的制备方法中,在分子筛合成过程中额外加入醇和金属源,并采用有机胺和有机模板剂配合使用,且采用两段变温醇-水热体系晶化,非常有利于微量的金属离子进入到分子筛骨架,得到的分子筛催化性能好。本发明制备的分子筛中含有微量金属元素,所述金属元素优选选自al、ag、co、ni、cu、zn、mn、pd、pt、cr、fe、la、au、ru、rh、y、ce、pt、rh、ti、zr、v、mo和w元素中的至少一种。
[0022]
与现有技术对比,本发明的有益效果包括:本发明成功制得了含有微量金属的mfi拓扑学结构硅分子筛,采用本发明提供的方法制得的分子筛就会具有较好催化性能。在环己酮肟气相贝克曼重排反应中,采用现有全硅分子筛作催化剂,较优情况下,环己酮肟转化率和己内酰胺选择性快速评价第6小时可以分别达到95%和94%以上,该数值基本上达到极限。本发明提供的分子筛中,金属元素的含量为5-100μg/g,将其应用于己内酰胺的生产中,可以提高环己酮肟的转化率和已内酰胺的选择性,提升气相重排新工艺技术的经济性。
附图说明
[0023]
图1是本发明实施例1所制得的mfi拓扑学结构硅分子筛的x射线衍射谱图;
[0024]
图2为本发明实施例1所制得的mfi拓扑学结构硅分子筛的扫描电镜照片。
具体实施方式
[0025]
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
[0026]
本发明第一方面提供一种mfi拓扑学结构硅分子筛,该分子筛含有硅元素、氧元素和金属元素,以分子筛的总量为基准,金属元素的含量为5-100μg/g;该分子筛的bet比表面积为400-500m
2
/g,所述金属元素选自过渡金属元素和iiia族金属元素中的至少一种。
[0027]
根据本发明的一种优选实施方式,该分子筛的bet比表面积为420-450m
2
/g。本发明提供的分子筛具有较高的bet比表面积。
[0028]
根据本发明的一种优选实施方式,该分子筛的外比表面积为35-60m
2
/g。
[0029]
本发明中,所述mfi拓扑学结构硅分子筛的bet比表面积和外比表面积可以采用n
2
吸附-脱附法进行测定,具体地,由美国micromeritics asap-2460型自动吸附仪测得,测试条件为:n
2
作吸附质,吸附温度为-196.15℃(液氮温度),在1.3pa、300℃下恒温脱气6h。
[0030]
根据本发明的一种优选实施方式,该分子筛的晶粒粒径为0.1-0.3μm,进一步优选为0.15-0.25μm。本发明中,分子筛的晶粒粒径采用扫描电镜获得。具体地,可以在日本hitachi公司s-4800场发射型扫描电镜上获得。
[0031]
根据本发明,优选地,金属元素的含量为6-90μg/g,进一步优选为20-60μg/g,例如可以为20μg/g、25μg/g、30μg/g、35μg/g、40μg/g、45μg/g、50μg/g、55μg/g、60μg/g,以及这些数值中的任意两个所构成的范围中的任意值。金属元素的含量过多,有可能使得分子筛路易斯酸增强,诱发不必要的副反应发生,不利于提高己内酰胺的选择性;而金属元素含量偏少,不利于分子筛寿命的延长和稳定性的提高。
[0032]
本发明中所述金属元素可以选自过渡金属元素和iiia族金属元素中的至少一种。进一步地,所述过渡金属元素选自第ib族、第iib族、第ivb族、第vb族、第vib族、第viib族和第viii族金属元素中的至少一种。
[0033]
根据本发明的一种优选实施方式,所述金属元素选自al、ag、co、ni、cu、zn、mn、pd、pt、cr、fe、la、au、ru、rh、y、ce、pt、rh、ti、zr、v、mo和w元素中的至少一种,进一步优选地,所述金属元素选自fe、ni、ti、pd、ce、al、cu、zr、pt和la元素中的至少一种。采用该种优选实施
方式,更有利于提高分子筛的催化性能。
[0034]
本发明所述金属元素含量使用美国pe(珀金埃尔默)公司7000dv型icp电感耦合等离子体原子发射光谱仪进行测定,测试条件为:用hf酸或王水溶解分子筛,使样品中的氧化硅和金属氧化物彻底溶解,在水溶液中测定金属离子含量。
[0035]
只要使用上述组成和结构的mfi拓扑学结构硅分子筛即可实现本发明的目的,本发明对含金属元素的mfi拓扑学结构硅分子筛的制备方法没有特别的限定。
[0036]
在本发明中,具体地,所述金属元素在分子筛中以金属离子形式存在。
[0037]
本发明第二方面提供一种mfi拓扑学结构硅分子筛的制备方法,该方法包括:
[0038]
(1)将硅源、有机胺、有机模板剂、金属源、有机醇和水混合,得到胶体混合物,其中,硅源、有机胺、有机模板剂、有机醇和水的用量摩尔比为1:(0.05-0.5):(0.05-0.5):(4-20):(5-100),硅源与金属源的用量质量比为(10000-200000):1,硅源以sio
2
计,金属源以金属元素计;
[0039]
(2)将所述胶体混合物进行两段变温醇-水热体系晶化,所述两段变温醇-水热体系晶化的条件包括:在40-70℃下晶化0.5-5天,然后在80-130℃下晶化0.5-5天;
[0040]
(3)将步骤(2)得到的晶化母液进行过滤、干燥和焙烧;
[0041]
所述金属选自过渡金属和iiia族金属中的至少一种。
[0042]
在本发明中,在无特殊说明情况下,所述用量摩尔比和用量质量比指的是物料进料(投料)时的用量摩尔比和用量质量比。
[0043]
根据本发明,优选地,所述硅源为有机硅源,进一步优选为有机硅酸酯,例如可以为通式是(or
1
)
4
si的有机硅酸酯,其中,r
1
为c1-c4的烷基。
[0044]
根据本发明,最优选地,所述硅源为正硅酸乙酯和/或正硅酸甲酯。
[0045]
根据本发明,优选地,所述有机胺选自脂肪胺类化合物中的至少一种。具体地,所述的脂肪胺类化合物的通式可以为(r
2
)
k
(nh
3-k
)
n
,r
2
为具有1-6个碳原子的烷基,n=1或2,k=1、2或,3进一步优选地,所述脂肪胺类化合物可以选自一正丙胺、二正丙胺、三正丙胺、乙胺、正丁胺、乙二胺和己二胺中的至少一种,最优选为三正丙胺。
[0046]
根据本发明,优选地,所述有机模板剂选自季铵碱类化合物,进一步优选为四丙基氢氧化铵和/或四乙基氢氧化铵。所述季铵碱类化合物可以为含有1-4个碳原子的烷基季铵碱类化合物,进一步优选为四丙基氢氧化铵和/或四乙基氢氧化铵。
[0047]
根据本发明提供的方法,所述金属和过渡金属的种类的选择如上文所述,在此不再赘述。
[0048]
根据本发明提供的方法,所述金属源为能够提供金属元素的各种含金属元素的化合物,优选地,所述金属源选自金属的硝酸盐、氯化物、硫酸盐、醋酸盐和酯类金属化合物中的至少一种。所述酯类金属化合物可以为钛酸乙酯和/或钛酸丁酯。
[0049]
根据本发明提供的方法,所述金属源优选为fe(no
3
)
3
、ni(no
3
)
2
、钛酸四丁酯、pd(no
3
)
2
、ce(no
3
)
4
、al(no
3
)
3
、cu(no
3
)
2
、zrocl
2
、h
2
ptcl
6
和la(oac)
3
中的至少一种。其中,“oac”代表乙酸根。上述金属源可以含有结晶水,也可以没有,本发明对此没有特别的限定。
[0050]
根据本发明提供的方法,所述有机醇可以为c1-c4的一元醇、二元醇中的至少一种,优选地,所述有机醇为乙醇和/或甲醇。
[0051]
本发明对步骤(1)所述混合的具体实施方式没有特别的限定,只要能够得到所述
胶体混合物即可。
[0052]
根据本发明的一种优选方式,所述硅源为正硅酸乙酯,所述有机醇为乙醇。本发明的发明人在研究过程中发现,正硅酸乙酯作为硅源和乙醇作为有机醇配合使用更有利于进一步提高制得的分子筛的催化性能。进一步优选地,步骤(1)所述混合包括:将乙醇、有机胺和有机模板剂进行第一混合,然后加入金属源和水,再加入正硅酸乙酯;或者,步骤(1)所述混合包括:将乙醇、有机胺和有机模板剂进行第一混合,然后依次加入水和正硅酸乙酯,再加入金属源。该种优选实施方式更有利于各物料的混合,同时更有利于发挥各物料的配合作用。
[0053]
根据本发明的一种具体实施方式,所述第一混合在搅拌条件下进行,对于搅拌的时间没有特别的限定,只要使得乙醇、有机胺和有机模板剂混合均匀即可。具体地,该方法还可以包括,在加入金属源和水之后,进行搅拌,然后加入所述正硅酸乙酯。
[0054]
根据本发明的一种具体实施方式,该方法还可以包括:在加入正硅酸乙酯之后,进行搅拌以得到所述胶体混合物。本发明对所述搅拌的条件没有特别的限定,以能够得到所述胶体混合物为准。例如,可以在常温(25℃)下搅拌2-6小时。
[0055]
根据本发明的另一种优选实施方式,所述硅源为正硅酸甲酯,所述有机醇为甲醇。本发明的发明人在研究过程中发现,正硅酸甲酯作为硅源和甲醇作为有机醇配合使用更有利于进一步提高制得的分子筛的催化性能。优选地,步骤(1)所述混合包括:将甲醇、有机胺和有机模板剂进行第二混合,然后加入金属源和水,再加入正硅酸甲酯;进一步优选地,所述正硅酸甲酯通过多次加入。采用该种优选实施方式,更有利于控制正硅酸甲酯的水解速度,进而使得制得的分子筛的催化性能得到进一步的提高。
[0056]
本发明对所述正硅酸甲酯通过多次加入的具体操作没有特别的限定,具体地,可以将所需量的正硅酸甲酯分为相等或不等的多份(优选为3-10份),然后将各份正硅酸甲酯间隔加入。本发明对所述间隔的时间没有特别的限定,所述间隔时间可以根据每次加入的正硅酸甲酯的量考虑,加入的正硅酸甲酯的量多,间隔时间可以适当延长,加入的正硅酸甲酯的量少,间隔时间可以适当地缩短。优选地,间隔时间可以为5-30min,各间隔时间可以相等也可以不等。本发明实施例中以正硅酸甲酯分为4批次,间隔时间为10min为例进行示例性说明,本发明并不限于此。
[0057]
根据本发明的一种优选实施方式,硅源、有机胺、有机模板剂、有机醇和水的用量摩尔比为1:(0.05-0.3):(0.05-0.3):(4-15):(15-50),进一步优选为1:(0.1-0.3):(0.1-0.2):(6-13):(20-40)。
[0058]
根据本发明,优选地,硅源与金属源的用量质量比为(10000-100000):1,进一步优选为(14000-50000):1。采用该种优选实施方式,进入分子筛骨架的适宜量的金属,更有利于提高分子筛的催化性能。
[0059]
根据本发明提供的方法,优选地,所述两段变温醇-水热体系晶化的条件包括:在50-65℃下晶化1-1.5天,然后在100-120℃下晶化1.5-2天。采用该种优选的水热体系晶化条件制得的分子筛具有更好的催化性能。
[0060]
根据本发明提供的方法,具体地,所述两段变温醇-水热体系晶化可以在密闭体系下,在自生压力下进行,例如在密闭反应釜中进行。
[0061]
本发明步骤(3)所述过滤可以为本领域常规使用的各种过滤方法,只要能够得到
固体产物即可。
[0062]
根据本发明的一种具体实施方式,本发明步骤(3)还包括在所述过滤之前进行的洗涤过程。本发明对所述洗涤过程使用的洗涤用剂没有特别的限定,例如可以为水。
[0063]
本发明对步骤(3)所述干燥的选择范围较宽,所述干燥的条件可以包括:温度为80-150℃,时间为2-36h。
[0064]
本发明对步骤(3)所述焙烧选择范围较宽,优选地,所述焙烧的条件包括:温度为400-600℃,时间为1-20h。
[0065]
根据本发明的一种优选实施方式,该方法还包括:在步骤(3)所述过滤之前,将所述晶化母液进行赶醇。优选地,所述赶醇的条件包括:温度为50-85℃,时间为1-12h。
[0066]
具体地,在工业上,因为醇中含有有机氧,其排放到废水里会带来环保问题,故需要进行赶醇操作。具体地,可以是待反应釜温度降低至可操作温度,打开反应釜,将反应釜升高至50-85℃,使得有机醇蒸发。具体地,在赶醇操作中,可以向反应釜中加入水,以维持反应釜液位。
[0067]
本发明所述“第一”和“第二”不起到限定作用,只是为了区分不同阶段进行的操作。
[0068]
本发明第三方面提供上述的制备方法制得的mfi拓扑学结构硅分子筛。采用本发明提供的制备方法成功地使得金属离子进入分子筛骨架结构,且制得的分子筛晶粒更细,更均匀,催化性能更好。采用本发明提供的制备方法制得的mfi拓扑学结构硅分子筛的具体结构和组成特征如上所述,在此不再赘述。
[0069]
本发明第四方面提供了上述mfi拓扑学结构硅分子筛在环己酮肟气相贝克曼重排反应中的应用。将本发明提供的mfi拓扑学结构硅分子筛作为催化剂用于环己酮肟气相贝克曼重排反应,可以提高环己酮肟的转化率和已内酰胺的选择性,有利于提升气相重排新工艺技术的经济性。
[0070]
根据本发明的一种优选实施方式,将上述mfi拓扑学结构硅分子筛应用于环己酮肟气相贝克曼重排反应之前,还包括对所述mfi拓扑学结构硅分子筛进行碱处理,具体地,所述碱处理包括:将mfi拓扑学结构硅分子筛与含氮化合物的碱性缓冲溶液接触,然后进行干燥。优选地,所述接触的条件包括:温度为80-100℃,压力为2-3kg/cm
2
,时间为1-3小时。进一步优选地,所述接触在搅拌条件下进行。本发明对所述搅拌的速度没有特别的限定,本领域技术人员可以根据实际情况进行适当的选择。
[0071]
根据本发明提供的应用,与含氮化合物的碱性缓冲溶液接触的过程可以进行重复操作。对于重复的次数本发明不做特别的限定,可以根据分子筛的性能确定,例如可以重复1-3次。
[0072]
优选地,所述含氮化合物的碱性缓冲溶液的ph值为8.5-13.5,进一步优选为11-11.5。
[0073]
优选地,所述含氮化合物的碱性缓冲溶液含有铵盐和碱,其溶剂可以为水。所述含氮化合物可以为铵盐,例如可以为硝酸铵和/或醋酸铵。所述碱可以选自氨水、四甲基氢氧化铵、四乙基氢氧化铵和四丙基氢氧化铵中的至少一种,优选为氨水。
[0074]
根据本发明的一种优选实施方式,所述铵盐的含量可以为0.5-20重量%;所述碱的含量可以为5-30重量%。
[0075]
本发明对所述干燥的条件没有特别的限定,可以按照本领域常规技术手段进行,所述干燥只要将水分除去即可,所述干燥的方法包括但不限于自然干燥、加热干燥、鼓风干燥,所述干燥的温度可以为100-120℃,干燥的时间可以为2-36小时。
[0076]
根据本发明,具体地,还可以包括:在所述干燥之前,将所述mfi拓扑学结构硅分子筛与含氮化合物的碱性缓冲溶液接触之后得到的物质依次进行洗涤、过滤。本发明对所述洗涤过程使用的洗涤用剂没有特别的限定,例如可以为水。具体地,所述洗涤过程可以为:洗涤至滤清液的ph为7-10.5。
[0077]
以下通过实施例对本发明进行详细描述。
[0078]
以下实施例中,所述金属元素含量使用美国pe(珀金埃尔默)公司7000dv型icp电感耦合等离子体原子发射光谱仪进行测定,测试条件为:用hf酸或王水溶解分子筛,使样品中的氧化硅和金属氧化物彻底溶解,在水溶液中测定金属离子含量。
[0079]
分子筛的外比表面积和bet比表面积由美国micromeritics asap-2460型自动吸附仪测得,测试条件为:n
2
作吸附质,吸附温度为-196.15℃(液氮温度),在1.3pa、300℃下恒温脱气6h。x射线衍射光谱由日本理学miniflex600型衍射仪录得,测试条件为:cu靶kα辐射,ni滤光片,管电压40kv,管电流40ma。
[0080]
样品的表面形态在日本hitachi公司s-4800型场发射扫描电镜上进行获得。
[0081]
以下实施例中洗涤均采用水进行,洗至过滤出来的洗涤水ph接近8-9。
[0082]
实施例1
[0083]
将435克95重量%的乙醇、29.2克三正丙胺和90克22.5wt%的四丙基氢氧化铵水溶液分别加入到2000ml烧杯中,搅拌,然后向烧杯中加入270克水和fe(no
3
)
3
·
9h
2
o,继续搅拌,再加入208克正硅酸乙酯,常温(25℃)下搅拌4小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:乙醇:水的用量摩尔比=1:0.2:0.1:9:20,硅源与fe
3+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升锈钢反应釜中,先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s1。
[0084]
对mfi拓扑学结构分子筛s1的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s1的x射线衍射谱图如图1所示,扫描电镜照片如图2所示。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s1晶粒大小均匀,粒径为0.15-0.25μm。
[0085]
实施例2
[0086]
将290克95重量%的乙醇、43.8克三正丙胺和90克22.5wt%的四丙基氢氧化铵水溶液分别加入到2000ml烧杯中,搅拌,然后向烧杯中加入450克水和ni(no
3
)
2
,继续搅拌,再加入208克正硅酸乙酯,常温(25℃)下搅拌4小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:乙醇:水的用量摩尔比=1:0.3:0.1:6:30,硅源与ni
2+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在50℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s2。
[0087]
对mfi拓扑学结构分子筛s2的组成和结构进行分析,结果列于表1中。mfi拓扑学结
构分子筛s2的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s2晶粒大小均匀,粒径为0.15-0.25μm。
[0088]
实施例3
[0089]
将290克95重量%的乙醇、43.8克三正丙胺和90克22.5wt%的四丙基氢氧化铵水溶液分别加入到2000ml烧杯中,搅拌,然后向烧杯中加入450克水,继续搅拌,再加入208克正硅酸乙酯,搅拌,最后加入钛酸四丁酯于常温(25℃)下搅拌4小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:乙醇:水的用量摩尔比=1:0.3:0.1:6:30,硅源与ti
4+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在65℃下醇-水热体系晶化1天,再在120℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s3。
[0090]
对mfi拓扑学结构分子筛s3的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s3的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s3晶粒大小均匀,粒径为0.15-0.25μm。
[0091]
实施例4
[0092]
将435克95重量%的乙醇、29.2克三正丙胺和90克22.5wt%的四丙基氢氧化铵水溶液分别加入到2000ml烧杯中,搅拌,然后向烧杯中加入270克水和pd(no
3
)
2
·
2h
2
o,继续搅拌,再加入208克正硅酸乙酯,常温(25℃)下搅拌4小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:乙醇:水的用量摩尔比=1:0.2:0.1:9:20,硅源与pd
2+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在50℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s4。
[0093]
对mfi拓扑学结构分子筛s4的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s4的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s4晶粒大小均匀,粒径为0.15-0.25μm。
[0094]
实施例5
[0095]
将435克95重量%的乙醇、29.2克三正丙胺和90克22.5wt%的四丙基氢氧化铵水溶液分别加入到2000ml烧杯中,搅拌,然后向烧杯中加入270克水和ce(no
3
)
4
·
7h
2
o,继续搅拌,再加入208克正硅酸乙酯,常温(25℃)下搅拌4小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:乙醇:水的用量摩尔比=1:0.2:0.1:9:20,硅源与ce
4+
的用量质量比为23500:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在60℃下醇-水热体系晶化1天,再在120℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s5。
[0096]
对mfi拓扑学结构分子筛s5的组成和结构进行分析,结果列于表1中。mfi拓扑学结
构分子筛s5的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s5晶粒大小均匀,粒径为0.15-0.25μm。
[0097]
实施例6
[0098]
将290克95重量%的乙醇、14.6克三正丙胺和136克22.5wt%的四丙基氢氧化铵水溶液分别加入到2000ml烧杯中,搅拌,然后向烧杯中加入330克水和al(no
3
)
3
·
9h
2
o,继续搅拌,再加入208克正硅酸乙酯,常温(25℃)下搅拌4小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:乙醇:水的用量摩尔比=1:0.1:0.15:6:25,硅源与al
3+
的用量质量比为23500:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s6。
[0099]
对mfi拓扑学结构分子筛s6的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s6的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s6晶粒大小均匀,粒径为0.15-0.25μm。
[0100]
实施例7
[0101]
在25℃恒温下将288克甲醇、29.2克三正丙胺和90克22.5%的四丙基氢氧化铵水溶液倒入2000毫升烧杯中,搅拌,然后向烧杯中加入290克水和cu(no
3
)
2
·
2.5h
2
o,继续搅拌10min,分四批次、每批次间隔10分钟,将152.2克正硅酸甲酯加入上述混合溶液中,搅拌60min,之后25℃恒温下搅拌3小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:甲醇:水的用量摩尔比=1:0.2:0.1:9:20,硅源与cu
2+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s7。
[0102]
对mfi拓扑学结构分子筛s7的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s7的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s7晶粒大小均匀,粒径为0.15-0.25μm。
[0103]
实施例8
[0104]
在25℃恒温下将288克甲醇、29.2克三正丙胺和90克22.5%的四丙基氢氧化铵水溶液倒入2000毫升烧杯中,搅拌,然后向烧杯中加入290克水和al(no
3
)
3
·
9h
2
o,继续搅拌10min,分四批次、每批次间隔10分钟,将152.2克正硅酸甲酯加入上述混合溶液中,搅拌60min,之后25℃恒温下搅拌3小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:甲醇:水的用量摩尔比=1:0.2:0.1:9:20,硅源与al
3+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。
得到mfi拓扑学结构分子筛s8。
[0105]
对mfi拓扑学结构分子筛s8的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s8的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s8晶粒大小均匀,粒径为0.15-0.25μm。
[0106]
实施例9
[0107]
在25℃恒温下将192克甲醇、43.8克三正丙胺和90克22.5%的四丙基氢氧化铵水溶液倒入2000毫升烧杯中,搅拌,然后向烧杯中加入450克水和zrocl
2
·
8h
2
o,继续搅拌10min,分四批次、每批次间隔10分钟,将152.2克正硅酸甲酯加入上述混合溶液中,搅拌60min,之后25℃恒温下搅拌3小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:甲醇:水的用量摩尔比=1:0.3:0.1:6:30,硅源与zr
4+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s9。
[0108]
对mfi拓扑学结构分子筛s9的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s9的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s9晶粒大小均匀,粒径为0.15-0.25μm。
[0109]
实施例10
[0110]
在25℃恒温下将288克甲醇、21.9克三正丙胺和135克22.5%的四丙基氢氧化铵水溶液倒入2000毫升烧杯中,搅拌,然后向烧杯中加入255克水和h
2
ptcl
6
·
6h
2
o,继续搅拌10min,分四批次、每批次间隔10分钟,将152.2克正硅酸甲酯加入上述混合溶液中,搅拌60min,之后25℃恒温下搅拌3小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化铵:甲醇:水的用量摩尔比=1:0.15:0.15:9:20,硅源与pt
4+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s10。
[0111]
对mfi拓扑学结构分子筛s10的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s10的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s10晶粒大小均匀,粒径为0.15-0.25μm。
[0112]
实施例11
[0113]
在25℃恒温下将192克甲醇、29.2克三正丙胺和90克22.5%的四丙基氢氧化铵水溶液倒入2000毫升烧杯中,搅拌,然后向烧杯中加入80克水和la(oac)
3
·
5h
2
o,继续搅拌10min,分四批次、每批次间隔10分钟,将152.2克正硅酸甲酯加入上述混合溶液中,搅拌60min,之后25℃恒温下搅拌3小时,形成胶体混合物,其中,硅源:三正丙胺:四丙基氢氧化
铵:甲醇:水的用量摩尔比=1:0.2:0.1:6:25,硅源与la
3+
的用量质量比为23600:1,其中,硅源以sio
2
计;将上述胶体混合物移入2000毫升不锈钢反应釜中,先在60℃下醇-水热体系晶化1天,再在100℃下醇-水热体系晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧6小时。得到mfi拓扑学结构分子筛s11。
[0114]
对mfi拓扑学结构分子筛s11的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s11的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s11晶粒大小均匀,粒径为0.15-0.25μm。
[0115]
实施例12
[0116]
按照实施例1的方法,不同的是,将三正丙胺替换为相同摩尔量的乙二胺。得到mfi拓扑学结构分子筛s12。
[0117]
对mfi拓扑学结构分子筛s12的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s12的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s12晶粒大小均匀,粒径为0.15-0.25μm。
[0118]
实施例13
[0119]
按照实施例1的方法,不同的是,将fe(no
3
)
3
·
9h
2
o替换为铝源(sb粉,氧化铝质量含量为70%,ti
4+
离子含量5μg/g),且硅源与al
3+
的用量质量比为15000:1。得到mfi拓扑学结构分子筛s13。
[0120]
对mfi拓扑学结构分子筛s13的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s13的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s13晶粒大小均匀,粒径为0.15-0.25μm。
[0121]
实施例14
[0122]
按照实施例1的方法,不同的是,将fe(no
3
)
3
·
9h
2
o替换为zrocl
2
·
8h
2
o,且硅源与zr
4+
的用量质量比为40000:1。得到mfi拓扑学结构分子筛s14。
[0123]
对mfi拓扑学结构分子筛s14的组成和结构进行分析,结果列于表1中。mfi拓扑学结构分子筛s14的x射线衍射谱图与图1相似,扫描电镜照片与图2相似。x射线衍射(xrd)谱图与microporous materials,vol 22,p637,1998上记载的mfi结构标准xrd谱图特征一致,这表明该分子筛具有mfi晶体结构。从扫描电镜照片可以看出,mfi拓扑学结构分子筛s14晶粒大小均匀,粒径为0.15-0.25μm。
[0124]
对比例1
[0125]
本对比例按照cn1124978c的方法合成硅分子筛。具体地:
[0126]
在室温25℃下,将139克正硅酸乙酯倒入1000毫升烧杯中,搅拌30分钟,用22.5%的四丙基氢氧化铵(tpaoh)溶液120克加入正硅酸乙酯中,室温25℃下搅拌水解2-3小时,升温到70-75℃,赶醇搅拌3-5小时,加水147克,形成溶胶,搅拌均匀,摩尔比tpaoh/sio
2
=
0.2,h
2
o/sio
2
=20,将上述混合物移入1000毫升不锈钢反应釜中,于170℃晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧5小时。得到分子筛d1。
[0127]
对分子筛d1的组成和结构进行分析,结果列于表1中。
[0128]
对比例2
[0129]
本对比例按照cn1124978c的方法合成硅分子筛。具体地:
[0130]
在室温25℃下,将139克正硅酸乙酯倒入1000毫升烧杯中,搅拌30分钟,用22.5%的四丙基氢氧化铵(tpaoh)溶液120克加入正硅酸乙酯中,室温25℃下搅拌水解5小时,加水147克,加乙醇(etoh)267克,搅拌形成溶胶,其中,摩尔比tpaoh/sio
2
=0.2,h
2
o/sio
2
=20,etoh/sio
2
=12.7,将上述混合物移入1000毫升不锈钢反应釜中,于110℃晶化2天,洗涤、过滤,120℃干燥24小时,550℃焙烧5小时。得到分子筛d2。
[0131]
对分子筛d2的组成和结构进行分析,结果列于表1中。分子筛d2的晶粒大小不均匀,粒径为0.1-0.4μm。
[0132]
对比例3
[0133]
按照实施例1的方法,不同的是,醇-水热体系晶化的条件为:100℃下醇-水热体系晶化3天。得到分子筛d3,对分子筛d3的组成和结构进行分析,结果列于表1中。分子筛d3的xrd结果显示,其特征衍射峰与分子筛s1相同,但特征衍射峰峰强度和峰高略有增加或增高。sem照片上显示分子筛晶粒相较于分子筛s1略有增大。
[0134]
对比例4
[0135]
按照实施例1的方法,不同的是,分子筛制备过程中不添加乙醇。得到分子筛d4,对分子筛d4的组成和结构进行分析,结果列于表1中。分子筛d4的xrd结果显示,其特征衍射峰与分子筛s1相同,但特征衍射峰峰强度和峰高略有增加或增高。sem照片上显示分子筛晶粒相较于分子筛s1略有增大。
[0136]
对比例5
[0137]
按照实施例1的方法,不同的是,分子筛制备过程中不添加fe(no
3
)
3
·
9h
2
o,即胶体混合物中不含有fe(no
3
)
3
·
9h
2
o。得到分子筛d5,对分子筛d5的组成和结构进行分析,结果列于表1中。
[0138]
表1
[0139][0140][0141]
试验例1
[0142]
本试验例1用于说明本发明提供的分子筛作为催化剂在环己酮肟气相贝克曼重排反应中的催化反应效果。具体地:
[0143]
将50克上述分子筛分别与500克氨水和硝酸铵组成的碱性缓冲溶液(其中,氨水与硝酸铵水溶液的重量比为3:2,ph值为11.35)加入到带压反应釜(kcf1-5型高压釜,烟台科
立化工设备有限公司)中,在80℃、2.3kg/cm
2
压力下搅拌2小时,然后洗涤、过滤、干燥,得到含不同金属元素的mfi结构分子筛的催化剂。
[0144]
反应装置为常压连续流动固定床,反应器内径为5毫米,催化剂的装填量0.36克,催化剂粒度20-60目。催化剂在装入反应管后,在常压、350℃的氮气气氛中预处理1小时。原料环己酮肟的浓度为35.7质量%,环己酮肟重量空速(whsv,进料中环己酮肟流量/反应器中催化剂重量)为16h-1
,溶剂为甲醇,反应温度为380℃,氮气流量为2.7升/小时,反应时间为6h。
[0145]
测试反应第6小时的环己酮肟转化率和己内酰胺的选择性,将反应产物通过水循环冷却后收集。采用毛细管气相色谱法测定产物组成,氢火焰检测器,测试条件为:汽化室温度250℃,检测室温度为230℃,柱温为程序升温,110℃恒温8分钟,15℃/min升到230℃再恒温14分钟。反应结果见表2。
[0146]
其中,
[0147]
环己酮肟转化率(mol%)=(100-反应产物中环己酮肟摩尔百分含量)/100
×
100%
[0148]
己内酰胺选择性(mol%)=反应产物中己内酰胺摩尔百分含量/(100-反应产物中环己酮肟摩尔百分含量)
×
100%
[0149]
甲基-ε-己内酰亚胺缩合物选择性(mol%)=反应产物中甲基-ε-己内酰亚胺缩合物摩尔百分含量/(100-反应产物中环己酮肟摩尔百分含量)
×
100%
[0150]
甲基-ε-己内酰亚胺缩合物水解会转化为己内酰胺和甲醇。
[0151]
表2
[0152][0153][0154]
注:表2中“cpl”代表己内酰胺,“amh”代表甲基-ε-己内酰亚胺缩合物。
[0155]
从表2结果可以看出,采用本发明提供的制备方法合成的硅分子筛,其环己酮肟的转化率可以达到99%以上,己内酰胺的总选择性(己内酰胺和甲基-ε-己内酰亚胺缩合物)
可以达到96.50%以上,优于现有技术中cn1124978c方法所合成的硅分子筛和对比例合成硅分子筛的结果。表明本发明提供的制备方法所得硅分子筛,在己内酰胺的生产中,可提高环己酮肟的转化率以及己内酰胺的选择性。
[0156]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips