
 商标分类
商标分类  商标转让
商标转让 
通过空间转录组技术分析牙周炎免疫微环境的方法与流程
 2021-01-08 11:01:18|
2021-01-08 11:01:18| 375|
375| 起点商标网
起点商标网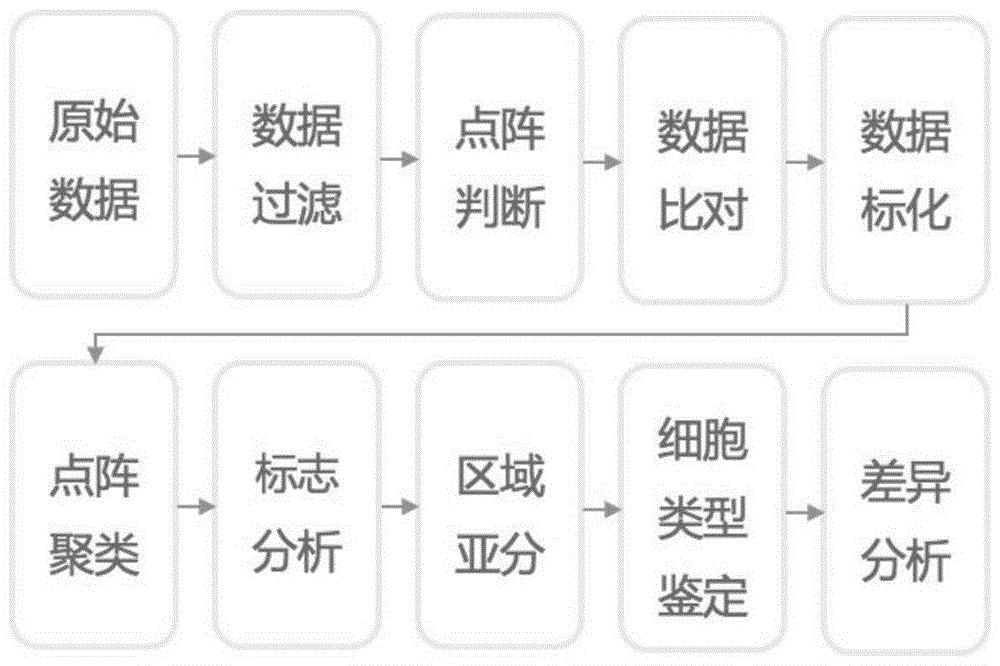
本发明属于口腔医学与生物信息技术领域,尤其涉及一种通过空间转录组技术分析牙周炎免疫微环境的方法。
背景技术:
牙周炎是以牙周组织破坏、牙槽骨吸收、牙齿松动为主要特点的一类慢性炎症性疾病,是最为常见的口腔炎性疾病之一。流行病学调查显示,全球牙周炎的患病率高达11.2%,且在40岁左右达患病高峰。
当机体牙周组织受到病原体或病原菌的刺激发生炎症时,即牙周炎发生时,释放的炎症因子将招募免疫细胞聚集于炎症病灶。然而,在攻击病原体或病原菌的同时,免疫细胞还会攻击机体自身正常组织细胞,而口腔作为有菌环境,长期处于带菌状态,因而这种炎症免疫攻击也将长期持续存在,产生大量炎症因子,对于机体自身正常组织的损伤较为严重,并干扰组织再生修复过程。
研究表明,牙周炎导致的骨组织丧失与炎症诱导的免疫反应间有着密切联系。这一过程不仅和单核巨噬细胞、成纤维细胞等介导的固有免疫反应相关,更与t细胞、b细胞所介导的适应性免疫反应关系密切。牙周炎免疫微环境主要是指牙周炎微环境中免疫细胞及免疫相关分子,针对牙周炎微环境中免疫环境的分类有助于了解免疫组成和免疫状态是如何影响炎症细胞和牙周炎治疗,但现有研究对于牙周炎的具体免疫调控机制尚不明确。因此,针对牙周炎免疫调控机制的研究具有重要现实意义与应用前景,亟需新思路与新方法对牙周炎免疫微环境进行深入探索研究。
针对rna测序,目前主要的测序方法包括群体转录组测序(bulkrna-seq)、单细胞测序(single-cellrna-seq)以及空间转录组测序。群体转录组测序是检测每个基因在大的细胞群体中的表达情况,主要用于比较基因组学,可用于比较不同物种的相同组织的基因表达差异;单细胞测序是测定每个基因在单个细胞中的表达情况,更具有微观性,相比前者能够检出混杂样品测序所无法得到的异质性信息;而空间转录组测序,则在单细胞测序的基础上,额外保留了细胞的空间位置信息,能将基因表达投影到现有空间信息,了解特定细胞与组织切片的相对位置关系。
技术实现要素:
本发明的目的在于提供一种通过空间转录组技术分析牙周炎免疫微环境的方法,旨在解决现有研究方法无法清楚分析和了解牙周炎免疫调控机制的问题。
本发明是这样实现的,一种通过空间转录组技术分析牙周炎免疫微环境的方法,该方法包括以下步骤:
(1)通过冰冻切片技术获得不同牙周炎患者及正常对照的牙周组织切片,
(2)通过空间转录组技术对每个组织样本进行测序,得到空间转录组数据;
(3)根据所述空间转录组数据进行标准数据分析,对所有检测到的牙周组织不同区域免疫微环境中的细胞种类构成进行分群、鉴定及有效定位,并对不同区域的原始基因的种类和表达情况进行进一步聚类与差异分析,以选出牙周炎免疫调控机制中的关键基因及相关分子信号通路。
优选地,在步骤(2)中,通过空间转录组技术对每个组织切片中被载玻片捕获区域内的寡核苷酸链捕获并标记唯一分子标识符umi和特殊空间条形码的mrna进行测序,得到空间转录组数据。
优选地,在步骤(3)中,所述根据所述空间转录组数据进行标准数据分析包括以下步骤:
对空间转录组数据采用fastp软件过滤获得可直接用于后续分析的测序数据;
采用barcode处理算法,将过滤得到的统计测序数据中所包含的cellbarcode信息及相对应的counts数进行统计,从而判断测序样本中实际检测到的点阵数,获得真实的空间转录组测序信息;
将测序数据中cellbarcode对应的reads比对到已知物种相对应的基因组,分析所测的未知序列与已知序列间的相似性与差异性,并获得与之比对的bam文件;
将上述包含有基因组比对后各种信息的bam文件进行转化,将文件中比对到同一基因上的单分子标签进行合并,并去除其中重复的umi序列,得到每个基因对应的umi数量,进而统计得到每个空间点阵所检测到的基因数,并在空间染色片中进行可视化展示。
优选地,在步骤(3)中,所述对所有检测到的牙周组织不同区域免疫微环境中的细胞种类构成进行分群、鉴定及有效定位包括以下步骤:
基于pca算法,将空间测序数据进行降维处理,得到二维/低维信息,利用t-sne算法和umap算法,将几千个空间点阵中具有相似性的spot进行聚类,进而得到聚类亚群(cluster),并在空间染色片中进行可视化展示;
通过对两两聚类簇间每个基因的log表达量进行welcht-tests,得到top10的差异基因作为marker基因候选,结合cellmarker数据库中已被验证过的marker基因,最终鉴定出每个cluster的marker基因。
优选地,在步骤(3)中,所述对不同区域的原始基因的种类和表达情况进行进一步聚类与差异分析包括以下步骤:
借助clusterprofiler软件包将上述聚类簇的marker基因与cellmarker数据库中的基因做富集分析,观察特定聚类簇富集于何种细胞类型,进而完成对细胞类型的鉴定;
将上述通过富集分析所得到的各聚类簇中具有明显差异表达的基因作为候选基因群,与各大数据库结合分析;
通过geneontology数据库对候选基因的细胞组件、分子功能、生物过程进行分析;
利用kegg数据库对候选基因进行pathwayanalysis,观察候选基因主要富集于哪些信号通路上;
将候选基因与disgenet数据库进行结合,通过富集分析观察候选基因与哪些疾病密切相关。
本发明克服现有技术的不足,提供一种通过空间转录组技术分析牙周炎免疫微环境的方法,本发明通过冰冻切片技术获得不同牙周炎患者及正常对照的牙周组织切片,进而借助空间转录组技术对每个组织样本进行测序,利用测序所得的空间转录组数据进行后续分析,再对所有检测到的牙周组织不同区域免疫微环境中的细胞种类构成进行分群、鉴定及有效定位,并对不同区域的原始基因的种类和表达情况进行进一步聚类与差异分析,以助于选出牙周炎免疫调控机制中的关键基因及相关分子信号通路。
相比于现有技术的缺点和不足,本发明具有以下有益效果:通过本发明方法可获得完整组织切片中具体空间位置的转录信息,有助于筛选出牙周炎免疫调控机制中的关键基因及相关分子信号通路,并为最终了解牙周炎的发生发展过程提供强力有效的数据支持。
附图说明
图1是本发明通过空间转录组技术分析牙周炎免疫微环境的方法的操作流程图;
图2是本发明实施例中牙周炎患者的牙周组织切片位置示意图;
图3是本发明实施例中counts数、点阵数、空间转录组测序信息的示例性展示图;
图4是本发明实施例中数据标化处理后的示例性展示图;
图5是本发明实施例中聚类亚群在空间染色片中进行可视化展示图;图5a为通过umap分析,将所有的spot依据相似性进行聚类,得到14个聚类簇(cluster),所有相同颜色的点(spot)构成一个聚类簇;图5b为依据每个spot先前保留的空间位置信息,将14个聚类亚群的所有spot对应的具体空间位置还原到切片上;
图6是本发明实施例中cxcl6基因在牙周组织切片不同部位的表达量;图6a为牙周炎组织切片图;图6b反映了cxcl6基因在牙周组织切片不同部位的表达量,图中点阵的颜色越深,则代表cxcl6基因在该处的表达量越高;其中,cxcl6在图6b右下角区域表达量最高,而对应左图的相同区域内有较多深染的炎症细胞的聚集,说明cxcl6基因可能是牙周炎相关基因。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
本发明公开了一种通过空间转录组技术分析牙周炎免疫微环境的方法,如图1所示,该方法包括以下步骤:
(1)通过冰冻切片技术获得不同牙周炎患者及正常对照的牙周组织切片
在步骤(1)中,临床收集健康对照、牙龈炎和牙周炎病人的牙周组织。所有受试者总体健康状况良好,在取材前3个月内未服用抗菌或抗炎药物。年龄相当的健康对照、牙龈炎和治疗前牙周炎病人的纳入均保证口腔内至少存在16颗牙并在取材前6个月内未经过牙周治疗。
1)健康对照纳入标准为:牙龈健康无炎症(探查不出血,且探诊深度<4mm),无临床附着丧失或骨丢失,由于义齿或正畸需要,在冠延长术中收集健康的牙周组织样本;
2)牙龈炎纳入标准:因牙位不正、拥挤或冠周炎需拔除的牙齿,存在牙龈炎症(探查出血,但无临床附着丧失或骨丢失),在牙拔除术时收集牙周组织样本(牙龈炎是牙周炎疾病发展早期,因此亦将其纳入测序样本);
3)牙周炎治疗前纳入标准:牙周炎需拔除的患牙(探查出血,且探诊深度>5mm,骨丢失>60%根长),在牙拔除术时收集牙周组织样本;
收集到的样本立即放置在含有αmem培养基的无菌管中,且在2小时内转移到实验室进行研究;
(2)异戊烷冻存:将采集的样本用预冷的异戊烷(冷冻剂)冻存,利于维持牙周组织的空间形态并保护rna的质量;
(3)oct包埋:将冷冻牙周组织放置于模具内,用预冷好的oct冰冻切片包埋剂将组织块完全包埋,包埋过程中应避免产生气泡。包埋剂有利于保留组织的完整结构,并在冷冻切片过程中提供组织的结构支撑,以增加组织连续性,减少皱折及碎裂,从而后续切片制备做好准备;
(4)切片制备:用刀片在包埋好的组织块表面切一个浅切口(约1mm深),避免切口过深损伤组织,再沿着切口对组织块进行切割,使切片厚度保持在10~20μm之间为宜,如图2所示。
(2)通过空间转录组技术对每个组织样本进行测序,得到空间转录组数据
步骤(2)包括以下具体步骤:
(i)数据过滤
为使数据更有效,本发明实施例需要对数据进行过滤处理。
采用fastp软件过滤掉原始数据(rawdata)中的低质量reads(测序片段)、未检测reads、包含接头的reads、等,从而获得可直接用于后续分析的cleanreads(cleandata),包括cellbarcode信息及基因计数(counts数)。
(ii)点阵判断
组织细胞释放出的mrna会迁移至测序芯片上的标记有特殊标签序列(cellbarcode)的spot(点阵)上。采用barcode处理算法,将过滤得到的统计测序数据(cleandata)中所包含的cellbarcode信息及相对应的基因计数counts数进行统计,counts数代表测序过程中测到相同的reads/测序片段出现的次数,即相应reads对应的丰度,反映了这一个基因转录的次数,也就是基因的表达量。根据该counts数判断测序样本中实际检测到的点阵数(spots),获得真实的空间转录组测序信息。结果示例如图3所示。
(iii)数据比对
将测序数据中cellbarcode对应的reads(受目前测序水平限制,基因组测序时需要先将基因组打断成dna片段,然后再建库测序,reads就是每次测序后对应的测序片段的读长/碱基序列的长度)比对到已知物种(人)相对应的基因组中。通过比对,能够发现所测的未知序列与已知序列间的相似性与差异性,并获得与之比对的bam文件(bam文件是sam文件的二进制版,存储了测序后map/比对到基因组后的各种信息,并极大地压缩了文件大小);
(iv)测序数据标化
对于测序数据,由于技术水平的差异,会出现测序深度、基因长度的不同。因此,为能将不同样本、不同基因的表达量进行比较,需要对原始测序数据进行标准化处理。
在本发明实施例中,数据标化处理的方法是将上述包含有基因组比对后各种信息的bam文件进行转化,将文件中比对到同一基因上的单分子标签(uniquemolecularidentifier,umi)进行合并,并去除其中重复的umi序列,得到每个基因对应的umi数量(每条mrna都标记有唯一分子标识符umi,可用于对mrna分子的计数),进而统计得到每个空间点阵(spot)所检测到的基因数(反映了一个spot当中有多少个基因的表达被检测到(一个细胞当中能表达的基因数量越多,则细胞的分化程度越低))及mrna分子数量。
对空间切片进行染色,若某点染色程度越深,则代表该点表达的基因数越多,可能反映位于该点细胞分化程度较低(与该点所包含的细胞种类数有关),例如如图4所示。
(3)根据所述空间转录组数据进行标准数据分析,对所有检测到的牙周组织不同区域免疫微环境中的细胞种类构成进行分群、鉴定及有效定位,并对不同区域的原始基因的种类和表达情况进行进一步聚类与差异分析,以选出牙周炎免疫调控机制中的关键基因及相关分子信号通路
本发明实施例中,步骤(3)包括以下具体步骤:
(v)点阵聚类
基于pca算法,将空间测序数据进行降维处理,得到二维/低维信息。再利用t-sne算法和umap算法,将几千个空间点阵(spot)中具有相似性的spot进行聚类,进而得到十几或二十几个的聚类亚群(cluster),并在空间染色片中进行可视化展示,结果如图5所示,图中,通过umap分析,将所有的spot依据相似性进行聚类,得到14个聚类簇(cluster),并将cluster对应的具体空间位置还原到切片上。
在本发明实施例中,此处分群的目的在于,基于t-sne算法和umap算法将几千个spot依据基因表达情况进行聚类,得到十几或二十几个的cluster,后期再结合公共数据库,根据聚类情况对cluster内的spot进行细胞种类鉴定与基因功能分析。
(vi)marker分析
点阵聚类后得到了不同的cluster,接下来就可以为每个cluster寻找marker基因。通过对两两聚类簇(cluster)间每个基因的log表达量进行welcht-tests,得到top10的差异基因作为marker基因候选,再结合cellmarker数据库中已被验证过的marker基因,最终鉴定出每个cluster的marker基因。以基因表达空间图的方式对marker基因的表达分布在切片上进行可视化展示,如图6所示,图6反映了cxcl6基因在牙周组织切片不同部位的表达量,图中点阵的颜色越深,则代表marker基因在该处的表达量越高。
通过对marker基因的鉴定,可用于后续判断或鉴定细胞类型。
(vii)细胞类型鉴定
借助clusterprofiler(r语言的一个软件包),将上述聚类簇(cluster)的marker基因与cellmarker数据库中的基因做富集分析,观察特定聚类簇富集于何种细胞类型,进而完成对细胞类型的鉴定。
(viii)差异分析
将上述通过富集分析所得到的各聚类簇中具有明显差异表达的基因作为候选基因群,与各大数据库结合分析。通过go(geneontology)数据库对候选基因的细胞组件、分子功能、生物过程进行分析;利用kegg数据库对候选基因进行pathwayanalysis,观察候选基因主要富集于哪些信号通路上;此外,将候选基因与disgenet数据库进行结合,通过富集分析观察候选基因与哪些疾病密切相关。综合以上各项结果,对差异表达基因的相关信息进行全方位的完整描述。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



