
 商标分类
商标分类  商标转让
商标转让 
一种蛋白质特征提取方法、装置及计算机设备与流程
 2021-01-08 11:01:10|
2021-01-08 11:01:10| 248|
248| 起点商标网
起点商标网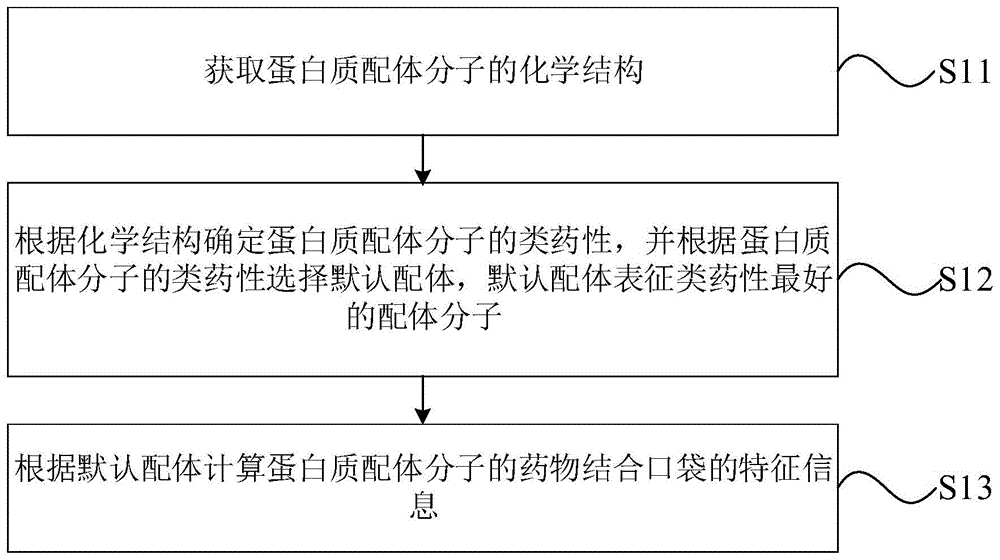
本发明涉及生物技术领域,具体涉及一种蛋白质特征提取方法、装置及计算机设备。
背景技术:
在计算机辅助药物设计中,基于结构的虚拟筛选技术主要依赖于生物大分子,如蛋白、核酸等结构的准确性。目前获取生物大分子结构的实验方法主要包括:x-射线晶体衍射技术、核磁共振技术、冷冻电镜技术等,这三种方法可信度较高,但是动辄需要数十亿的设备和数月的时间进行结构解析,解析获得的结构一般会存到世界蛋白质数据库(worldwideproteindatabank,wwpdb)中。由于早期的实验数据精度不高,或解析出的结构无法满足一定的药物研发需求,因此,所有的生物大分子结构都需要进行合理的预处理才可以进行后续的研究工作。
相关技术中,采用传统软件,如薛定谔分子模拟软件
技术实现要素:
因此,本发明要解决的技术问题在于克服现有技术中需要用户手动处理蛋白质,浪费人力和时间的缺陷,从而提供一种蛋白质特征提取方法、装置及计算机设备。
根据第一方面,本发明实施例公开了一种蛋白质特征提取方法,包括如下步骤:获取蛋白质配体分子的化学结构;根据所述化学结构确定所述蛋白质配体分子的类药性,并根据所述蛋白质配体分子的类药性选择默认配体,所述默认配体表征类药性最好的配体分子;根据所述默认配体计算所述蛋白质配体分子的药物结合口袋的特征信息。
可选地,所述获取蛋白质配体分子的化学结构,包括:获取所述蛋白质配体分子对应的蛋白质在蛋白质数据库中的索引信息;根据所述索引信息在所述蛋白质数据库中获取所述蛋白质的结构文件;在所述结构文件中,得到所述蛋白质配体分子的化学结构。
可选地,所述根据所述化学结构确定所述蛋白质配体分子的类药性,包括:获取所述化学结构的目标属性特征;根据所述目标属性特征和预设的属性特征评估值确定所述蛋白质配体分子的类药性。
可选地,所述根据所述默认配体计算所述蛋白质配体分子的药物结合口袋的特征信息,包括:获取所述默认配体在预设坐标系中的各个方向坐标的最大值和最小值,得到初始药物结合口袋;根据所述各个方向坐标的最大值和最小值,构建所述初始药物结合口袋对应空间范围,对所述空间范围进行扩展处理,得到所述药物结合口袋;根据所述药物结合口袋的各个方向坐标的最大值和最小值计算所述蛋白质配体分子的药物结合口袋的特征信息。
可选地,所述方法还包括:获取所述蛋白质配体分子对应的蛋白质的结构文件;对所述蛋白质的结构文件中的氨基酸进行补全处理;对补全后的氨基酸进行突变,得到包含标准氨基酸的蛋白质结构;去除所述包含标准氨基酸的蛋白质结构中的非药物小分子;基于预设的ph值对去除了所述非药物小分子的蛋白质结构进行加氢操作;对加氢操作后的蛋白质结构进行能量最小化处理,得到蛋白质的结构文件。
可选地,在对加氢操作后的蛋白质结构进行能量最小化处理,得到目标蛋白质的结构文件之后,所述方法还包括:根据数据存储条件,将所述蛋白质的结构文件转换为预设格式存储到对应的数据库中。
根据第二方面,本发明实施例还公开了一种蛋白质特征提取装置,包括:第一获取模块,用于获取蛋白质配体分子的化学结构;选择模块,用于根据所述化学结构确定所述蛋白质配体分子的类药性,并根据所述蛋白质配体分子的类药性选择默认配体,所述默认配体表征类药性最好的配体分子;第一计算模块,用于根据所述默认配体计算所述蛋白质配体分子的药物结合口袋的特征信息。
可选地,所述第一获取模块包括:第一获取子模块,用于获取所述蛋白质配体分子对应的蛋白质在蛋白质数据库中的索引信息;第二获取模块,用于根据所述索引信息在所述蛋白质数据库中获取所述蛋白质的结构文件;第三获取模块,用于在所述结构文件中,得到所述蛋白质配体分子的化学结构。
根据第三方面,本发明实施例还公开了一种计算机设备,包括:至少一个处理器;以及与所述至少一个处理器通信连接的存储器;其中,所述存储器存储有可被所述至少一个处理器执行的指令,所述指令被所述至少一个处理器执行,以使所述至少一个处理器执行如第一方面或第一方面任一可选实施方式所述的蛋白质特征提取方法的步骤。
根据第四方面,本发明实施例还公开了一种计算机可读存储介质,其上存储有计算机程序,所述计算机程序被处理器执行时实现如第一方面或第一方面任一可选实施方式所述的蛋白质特征提取方法的步骤。
本发明技术方案,具有如下优点:
本发明提供的蛋白质特征提取方法及装置,通过获取蛋白质配体分子的化学结构,根据化学结构确定蛋白质配体分子的类药性,并根据蛋白质配体分子的类药性选择默认配体,默认配体表征类药性最好的配体分子,根据默认配体计算蛋白质配体分子的药物结合口袋的特征信息。本发明通过对从蛋白质数据库获取到的蛋白质配体分子的化学结构进行配体处理,可快速获得蛋白质特征,便于后续进行计算机辅助药物设计时的虚拟筛选,操作简单,不需要依赖有经验的操作人员进行手动操作,减少人力和时间消耗。
附图说明
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例中蛋白质特征提取方法的一个具体示例的流程图;
图2为本发明实施例中蛋白质特征提取装置的一个具体示例的原理框图;
图3为本发明实施例中计算机设备的一个具体示例图。
具体实施方式
下面将结合附图对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
在本发明的描述中,需要说明的是,术语“中心”、“上”、“下”、“左”、“右”、“竖直”、“水平”、“内”、“外”等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本发明和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。此外,术语“第一”、“第二”、“第三”仅用于描述目的,而不能理解为指示或暗示相对重要性。
在本发明的描述中,需要说明的是,除非另有明确的规定和限定,术语“安装”、“相连”、“连接”应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或一体地连接;可以是机械连接,也可以是电连接;可以是直接相连,也可以通过中间媒介间接相连,还可以是两个元件内部的连通,可以是无线连接,也可以是有线连接。对于本领域的普通技术人员而言,可以具体情况理解上述术语在本发明中的具体含义。
此外,下面所描述的本发明不同实施方式中所涉及的技术特征只要彼此之间未构成冲突就可以相互结合。
在计算机辅助药物设计中,基于结构的虚拟筛选技术主要依赖于生物大分子,如蛋白质、核酸等结构的准确性,而虚拟筛选技术主要基于蛋白质配体分子的药物结合口袋信息和蛋白质的结构进行筛选,而由于早期的实验数据精度不高,提取存储在蛋白质数据库中的蛋白质结构无法满足药物研发需求,因此本发明实施例公开了一种蛋白质特征提取方法以对蛋白质结构进行处理,便于进行后续的研发工作,如图1所示,包括如下步骤:
s11:获取蛋白质配体分子的化学结构。
示例性地,该蛋白质配体分子为可以与蛋白质结合的任何分子,蛋白质配体分子与蛋白质的结合方式主要有环区结合以及铰链式结合。该蛋白质配体分子的化学结构的获取可以直接从蛋白质数据库获取,也可以从蛋白质的结构文件中摘取,本发明实施例对该蛋白质配体分子的化学结构的获取方式不作限定,本领域技术人员可以根据实际情况选择。
s12:根据化学结构确定蛋白质配体分子的类药性,并根据蛋白质配体分子的类药性选择默认配体,默认配体表征类药性最好的配体分子。
示例性地,类药性指的是蛋白质配体分子与已知药物的相似性。具有类药性的化合物并不是药物,但是具有成为药物的可能。根据化学结构确定蛋白质配体分子的类药性可以采用排除法,例如可以基于蛋白质配体分子的分子特性会导致毒性和降低生物利用度等进行排除,也可以采用模仿已知药物的方法,即利用已知药物化学结构中有利的特征对蛋白质配体分子进行评估,也可以采用定量类药性评估方法,本发明实施例对该蛋白质配体分子的类药性的确定方法不作具体限定,本领域技术人员可以根据实际情况设定。
根据对各个蛋白质配体分子的类药性进行比较,将类药性最好的蛋白质配体分子选择出来,得到默认配体,对该默认配体进行处理。
s13:根据默认配体计算蛋白质配体分子的药物结合口袋的特征信息。
示例性地,该药物结合口袋为配体可能结合的区域,其形状可能通常为口袋状,也可以为其他形状,例如,管道状、凹槽状等。该药物结合口袋的特征信息包括药物结合口袋的中心坐标以及药物结合口袋的长宽高。这几个数据可代表该药物结合口袋。根据默认配体计算蛋白质配体分子的药物结合口袋的特征信息可以为根据默认配体在预设坐标系中各个方向坐标的最大值和最小值计算得到。
本发明提供的蛋白质特征提取方法,通过获取蛋白质配体分子的化学结构,根据化学结构确定蛋白质配体分子的类药性,并根据蛋白质配体分子的类药性选择默认配体,默认配体表征类药性最好的配体分子,根据默认配体计算蛋白质配体分子的药物结合口袋的特征信息。本发明通过对从蛋白质数据库获取到的蛋白质配体分子的化学结构进行配体处理,可快速获得蛋白质特征,便于后续进行计算机辅助药物设计时的虚拟筛选,操作简单,不需要依赖有经验的操作人员进行手动操作,减少人力和时间消耗。
作为本发明实施例一个可选实施方式,上述步骤s11包括:
首先,获取蛋白质配体分子对应的蛋白质在蛋白质数据库中的索引信息。
示例性地,该蛋白质数据库可以为结构生物信息学研究合作组织蛋白质数据库(researchcollaborationforstructuralbioinformaticsproteindatabank,rcsbpdb)、欧洲蛋白质数据库(proteindatabankineurope,pdbe)、日本蛋白质数据库(proteindatabankinjapan,pdbj),这些数据库均存储有蛋白质的结构文件,它们提供了不同的接口供不同的服务器使用。该索引信息可以为蛋白质的pdb编号。该蛋白质的索引信息可以直接在相关搜索引擎上输入蛋白质名称得到。
其次,根据索引信息在蛋白质数据库中获取蛋白质的结构文件。
示例性地,将该索引信息输入到处理器,处理器会根据索引信息直接从对应的蛋白质数据库调用蛋白质的结构文件,该结构文件是.pdb格式的文件。
再次,在结构文件中,得到蛋白质配体分子的化学结构。
示例性地,每个蛋白质存在零到多个蛋白质配体分子,本实施例仅考虑存在蛋白质配体分子的情况。在上述结构文件中,得到蛋白质配体分子的化学结构可以为当蛋白质存在蛋白质配体分子时,从蛋白质数据库中获取蛋白质配体分子的索引信息,该蛋白质配体分子的索引信息可以为(chemicalcomponentdictionary,ccd),根据该ccd编号从蛋白质的结构文件中摘取蛋白质配体分子的化学结构。
通过实施本发明,用户只需要提供需要处理的蛋白质的pdb编号即可自动获取到蛋白质配体分子的结构,无需进行其他额外操作,节约人力资源。同时本发明实施例链接了多个不同侧重的pdb数据库,将多方数据进行整合,可获取最新的蛋白质信息。
作为本实施例一个可选实施方式,上述s12包括:
首先,获取化学结构的目标属性特征。
示例性地,该目标属性特征可以包括分子量、脂水分配系数(logp)、拓扑表面积、氢键受体/供体数量,芳香环数量、可旋转键数量、毒性化学片段数等,这些目标特征属性可以直接从蛋白质配体分子的化学结构获得。
其次,根据目标属性特征和预设的属性特征评估值确定蛋白质配体分子的类药性。
示例性地,该预设的属性评估值可以为根据专家经验提前设置好存储在处理器中的,例如,分子量的预设评估值为300个-500个;脂水分配系数的预设评估值为5;氢键受体的预设评估值为5个;氢键配体的预设评估值为5个-10个,同理,其他的属性特征也会设置相应的预设评估值。根据目标属性特征是否在其对应的预设的属性特征评估值范围确定蛋白质配体分子的类药性,相比于其他类药性计算方法只能得到是否具有类药性的结果,本实施例采用的定量类药性评估方法综合考虑了多种属性特征,不仅仅得到是否有类药性,还可以得到不同蛋白质配体分子的类药性大小,得到的蛋白质配体分子的类药性更加准确。
作为本实施例一个可选实施方式,上述s13包括:
首先,获取默认配体在预设坐标系中的各个方向坐标的最大值和最小值,得到初始药物结合口袋。
示例性地,该预设坐标系可以为提前以统一标准设置的立体坐标系。各个方向坐标x,y,z的最大值和最小值均可以从蛋白质的结构文件中获取到,或者从蛋白质数据库中获取。将各个方向坐标的最大值和最小值的围成的区域作为初始药物结合口袋的空间范围。
其次,根据各个方向坐标的最大值和最小值,构建初始药物结合口袋对应空间范围,对空间范围进行扩展处理,得到药物结合口袋。
示例性地,将各个方向坐标的最大值和最小值围成的区域作为初始药物结合口袋对应的空间范围,对空间范围进行扩展处理可以为将该空间范围均扩展预设距离,得到完整的药物结合口袋。在本发明实施例中,该预设距离可以为5埃。
再次,根据药物结合口袋的各个方向坐标的最大值和最小值计算蛋白质配体分子的药物结合口袋的特征信息。
示例性地,根据药物结合口袋的x,y,z方向的最大值和最小值计算中心坐标c_x,c_y,c_z的方式可以为求各个方向最大值和最小值的平均值;根据药物结合口袋的x,y,z方向的最大值和最小值计算药物结合口袋的长宽高dim_x,dim_y,dim_z可以为求各个方向最大值和最小值的差。将这6个数记录到数据库,即可代表该药物结合口袋。
作为本实施例一个可选实施方式,该蛋白质特征提取方法还包括:
首先,获取蛋白质配体分子对应的蛋白质的结构文件。该蛋白质的结构文件的获取方式参见上述相关描述,在此不再赘述。
其次,对蛋白质的结构文件中的氨基酸进行补全处理。
示例性地,根据蛋白质的结构文件中的残基序列(seqres)的序列信息与预设数据库中的标准氨基酸序列信息进行对比,将氨基酸结构的缺失残基进行补充。
再次,对补全后的氨基酸进行突变,得到包含标准氨基酸的蛋白质结构。
示例性地,补全后的氨基酸可能是非标准氨基酸,使用openmm-pdbfixer函数对补全后的氨基酸进行突变,具体地,将该氨基酸与预设数据库中的标准氨基酸序列进行对比,将补全后的氨基酸的残基的侧链原子坐标删除,再将氨基酸编号改为突变后的氨基酸的编号。
再次,去除包含标准氨基酸的蛋白质结构中的非药物小分子。该非药物小分子可以包括:水分子、其他溶剂和一些杂原子。为了保证蛋白质结构的准确性,直接将包含标准氨基酸的蛋白质结构中的非药物小分子删除。
再次,基于预设的ph值对去除了非药物小分子的蛋白质结构进行加氢操作。
示例性地,该预设的ph值可以为7.4,基于预设的ph值对去除了非药物小分子的蛋白质结构进行加氢操作可以为使用openmm.app.modeller.addhydrogens函数按照该预设的ph值添加合理的氢原子。例如,碱性氨基酸(精氨酸、赖氨酸、组氨酸)、酸性氨基酸(谷氨酸、天冬氨酸),均可以使用标准酸度系数(pka)与预设的ph值判断其质子化状态,根据其质子化状态按照openmm存储的氨基酸拓扑结构对氨基酸进行氢原子添加。
再次,对加氢操作后的蛋白质结构进行能量最小化处理,得到蛋白质的结构文件。
示例性地,在上述处理过程中可能会引入一些不合理的张力,使用openmm.localenergyminimizer.minimize函数进行能量最小化处理,该能量最小化方法一般为最陡梯度下降法或共轭梯度下降法,梯度求解方法为给定分子力场(charm36力场)下的势能对坐标的微分。
作为本实施例一个可选实施方式,在上述对加氢操作后的蛋白质结构进行能量最小化处理,得到目标蛋白质的结构文件之后,该蛋白质特征提取方法还包括:
根据数据存储条件,将蛋白质的结构文件转换为预设格式存储到对应的数据库中。
示例性地,该数据存储条件可以为可存储于对应数据库的化学结构格式。该预设格式可以包括:sdf格式、mol2格式、pdb格式等。该对应的数据库可以为关系型数据库管理系统(mysql),该数据库为离线数据库,便于其他服务器随时进行调用。调用时可以使用蛋白质的pdb编号,也可以使用蛋白质的uniprotaccession编号。
本发明实施例还公开了一种蛋白质特征提取装置,如图2所示,包括:
第一获取模块21,用于获取蛋白质配体分子的化学结构;具体实现方式见实施例中步骤s11的相关描述,在此不再赘述。
选择模块22,用于根据化学结构确定蛋白质配体分子的类药性,并根据蛋白质配体分子的类药性选择默认配体,默认配体表征类药性最好的配体分子;具体实现方式见实施例中步骤s12的相关描述,在此不再赘述。
第一计算模块23,用于根据默认配体计算蛋白质配体分子的药物结合口袋的特征信息。具体实现方式见实施例中步骤s13的相关描述,在此不再赘述。
本发明提供的蛋白质特征提取装置,通过获取蛋白质配体分子的化学结构,根据化学结构确定蛋白质配体分子的类药性,并根据蛋白质配体分子的类药性选择默认配体,默认配体表征类药性最好的配体分子,根据默认配体计算蛋白质配体分子的药物结合口袋的特征信息。本发明通过对从蛋白质数据库获取到的蛋白质配体分子的化学结构进行配体处理,可快速获得蛋白质特征,便于后续进行计算机辅助药物设计时的虚拟筛选,操作简单,不需要依赖有经验的操作人员进行手动操作,减少人力和时间消耗。
作为本发明一个可选实施方式,上述第一获取模块21包括:
第一获取子模块,用于获取蛋白质配体分子对应的蛋白质在蛋白质数据库中的索引信息;具体实现方式见实施例中对应的步骤,在此不再赘述。
第二获取模块,用于根据索引信息在蛋白质数据库中获取蛋白质的结构文件;具体实现方式见实施例中对应的步骤,在此不再赘述。
第三获取模块,用于在结构文件中,得到蛋白质配体分子的化学结构。具体实现方式见实施例中对应的步骤,在此不再赘述。
作为本发明实施例一个可选实施方式,上述选择模块22包括:
第四获取模块,用于获取化学结构的目标属性特征;具体实现方式见实施例中对应的步骤,在此不再赘述。
第二计算模块,用于根据目标属性特征和预设的属性特征评估值确定蛋白质配体分子的类药性。具体实现方式见实施例中对应的步骤,在此不再赘述。
作为本发明实施例一个可选实施方式,上述第一计算模块23包括:
第五获取模块,用于获取默认配体在预设坐标系中的各个方向坐标的最大值和最小值,得到初始药物结合口袋;具体实现方式见实施例中对应的步骤,在此不再赘述。
第一处理模块,用于根据各个方向坐标的最大值和最小值,构建初始药物结合口袋对应空间范围,对空间范围进行扩展处理,得到药物结合口袋;具体实现方式见实施例中对应的步骤,在此不再赘述。
第一计算子模块,用于根据药物结合口袋的各个方向坐标的最大值和最小值计算蛋白质配体分子的药物结合口袋的特征信息。具体实现方式见实施例中对应的步骤,在此不再赘述。
作为本发明实施例一个可选实施方式,该蛋白质特征提取装置还包括:
第二获取子模块,用于获取蛋白质配体分子对应的蛋白质的结构文件;具体实现方式见实施例中对应的步骤,在此不再赘述。
第二处理模块,用于对蛋白质的结构文件中的氨基酸进行补全处理;具体实现方式见实施例中对应的步骤,在此不再赘述。
突变模块,用于对补全后的氨基酸进行突变,得到包含标准氨基酸的蛋白质结构;具体实现方式见实施例中对应的步骤,在此不再赘述。
去除模块,用于去除包含标准氨基酸的蛋白质结构中的非药物小分子;具体实现方式见实施例中对应的步骤,在此不再赘述。
加氢操作模块,用于基于预设的ph值对去除了非药物小分子的蛋白质结构进行加氢操作;具体实现方式见实施例中对应的步骤,在此不再赘述。
第三处理模块,用于对加氢操作后的蛋白质结构进行能量最小化处理,得到蛋白质的结构文件。具体实现方式见实施例中对应的步骤,在此不再赘述。
作为本发明实施例一个可选实施方式,该蛋白质特征提取装置还包括:
存储模块,用于根据数据存储条件,将蛋白质的结构文件转换为预设格式存储到对应的数据库中。具体实现方式见实施例中对应的步骤,在此不再赘述。
本发明实施例还提供了一种计算机设备,如图3所示,该计算机设备可以包括处理器31和存储器32,其中处理器31和存储器32可以通过总线或者其他方式连接,图3中以通过总线连接为例。
处理器31可以为中央处理器(centralprocessingunit,cpu)。处理器31还可以为其他通用处理器、数字信号处理器(digitalsignalprocessor,dsp)、专用集成电路(applicationspecificintegratedcircuit,asic)、现场可编程门阵列(field-programmablegatearray,fpga)或者其他可编程逻辑器件、分立门或者晶体管逻辑器件、分立硬件组件等芯片,或者上述各类芯片的组合。
存储器32作为一种非暂态计算机可读存储介质,可用于存储非暂态软件程序、非暂态计算机可执行程序以及模块,如本发明实施例中的蛋白质特征提取方法对应的程序指令/模块(例如,图2所示的第一获取模块21、选择模块22和第一计算模块23)。处理器31通过运行存储在存储器32中的非暂态软件程序、指令以及模块,从而执行处理器的各种功能应用以及数据处理,即实现上述方法实施例中的蛋白质特征提取方法。
存储器32可以包括存储程序区和存储数据区,其中,存储程序区可存储操作系统、至少一个功能所需要的应用程序;存储数据区可存储处理器31所创建的数据等。此外,存储器32可以包括高速随机存取存储器,还可以包括非暂态存储器,例如至少一个磁盘存储器件、闪存器件、或其他非暂态固态存储器件。在一些实施例中,存储器32可选包括相对于处理器31远程设置的存储器,这些远程存储器可以通过网络连接至处理器31。上述网络的实例包括但不限于互联网、企业内部网、局域网、移动通信网及其组合。
所述一个或者多个模块存储在所述存储器32中,当被所述处理器31执行时,执行如图1所示实施例中的蛋白质特征提取方法。
上述计算机设备具体细节可以对应参阅图1所示的实施例中对应的相关描述和效果进行理解,此处不再赘述。
本领域技术人员可以理解,实现上述实施例方法中的全部或部分流程,是可以通过计算机程序来指令相关的硬件来完成,所述的程序可存储于一计算机可读取存储介质中,该程序在执行时,可包括如上述各方法的实施例的流程。其中,所述存储介质可为磁碟、光盘、只读存储记忆体(read-onlymemory,rom)、随机存储记忆体(randomaccessmemory,ram)、快闪存储器(flashmemory)、硬盘(harddiskdrive,缩写:hdd)或固态硬盘(solid-statedrive,ssd)等;所述存储介质还可以包括上述种类的存储器的组合。
虽然结合附图描述了本发明的实施例,但是本领域技术人员可以在不脱离本发明的精神和范围的情况下作出各种修改和变型,这样的修改和变型均落入由所附权利要求所限定的范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



