
 商标分类
商标分类  商标转让
商标转让 
一种抗肿瘤靶向药物缓释载体、制剂及其制备方法与流程
 2021-01-08 11:01:53|
2021-01-08 11:01:53| 363|
363| 起点商标网
起点商标网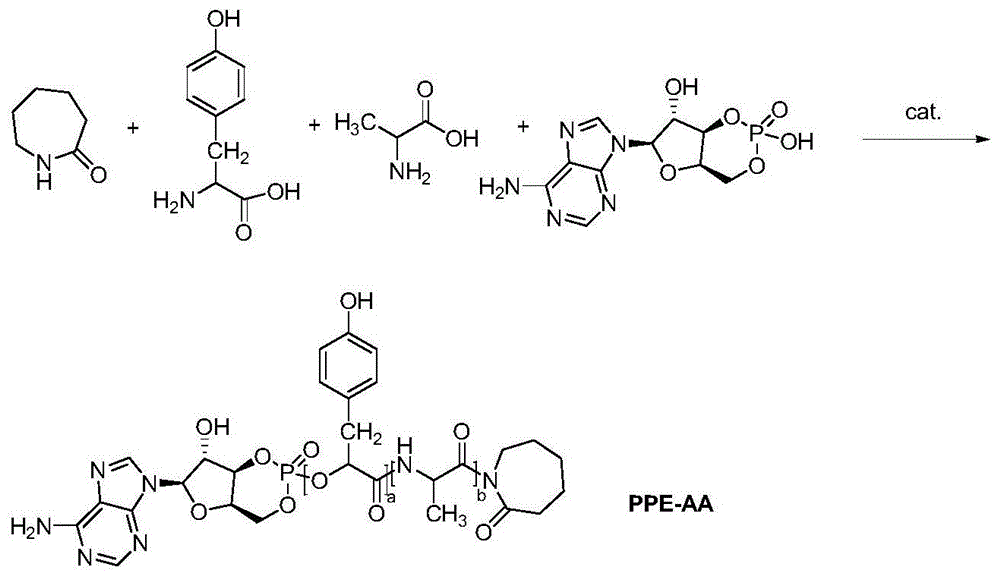
本发明涉及缓释药物
技术领域:
,具体涉及一种抗肿瘤靶向药物缓释载体、制剂及其制备方法。
背景技术:
:抗肿瘤药物毒性大,易在体内被水解,应用有效的药物量后患者的血药水平上升,达到极大值后却又迅速下降趋于零,导致药物还没有到达肿瘤细胞组织,药效已所剩无几。抗肿瘤药物缓控释制剂的出现改善了上述情况,抗肿瘤药物缓控释制剂克服了传统抗肿瘤药半衰期短、难以长时间维持有效浓度以及缺乏靶向性等缺点,近年来,出现了包括凝胶、微球、纳米微粒、植入剂等多种缓控释剂型,可经多种途径给药应用于肿瘤治疗。在临床治疗中,药物的低毒和高效一直是人们研究的重点,化疗是临床上治疗肿瘤的主要手段,但是目前传统的化疗药物普通剂型缺乏靶向性,在杀伤肿瘤细胞的同时也会对正常细胞产生毒性,引起的毒副作用使患者不能耐受。因此,构建靶向肿瘤组织的药物传递系统是解决肿瘤化疗问题的有效途径。近年来,叶酸受体作为抗肿瘤药物的靶点受到了极大的关注,成为新型抗肿瘤药物研究的热点之一。叶酸是小分子量维生素,相对于单分子抗体等蛋白质,具有结构稳定、价格低廉、无免疫原性等特点。研究发现,叶酸受体在绝大多数恶性肿瘤细胞膜内都过度表达而正常细胞很少表达甚至不表达,而且叶酸与叶酸受体结合力强,能被高效介导进入肿瘤细胞,基于这种特性,可以实现叶酸-药物偶联到药物载体上,实现主动靶向运输。叶酸受体介导的靶向给药系统常以叶酸或叶酸类似物为载体,将放射性核素、抗肿瘤药物、基因药物与之偶联,实现靶向输送药物的作用。由于多数叶酸复合物的体积较大,不易达到肿瘤细胞和被肿瘤细胞摄取,故常用纳米级的叶酸偶联物作为叶酸受体介导的靶向给药载体,如脂质体、胶束、纳米粒、乳剂、树枝状聚合物、超分子囊泡状聚合物等。缓释微球是指药物溶解或分散在高分子材料基质中形成的球状实体,在现有的众多缓释制剂中,微球制剂由于其特有的优点诸如给药后微球降解逐渐释放药物、可避免出现血药浓度峰谷现象减小毒副作用、大大减少治疗周期的给药剂量、可提高药物的生物利用度与病人的顺应性等,从而逐渐成为现代药剂学中热门的研究内容。利用缓释微球开发新型的给药系统逐渐成为科学界及工业界关注的焦点。技术实现要素:本发明的目的在于提出一种抗肿瘤靶向药物缓释载体、制剂及其制备方法,提高了药物的选择性和定位的准确性,经酶水解后释放出抗肿瘤药物,高效杀灭癌细胞,提高药效,缓释制剂可以有效避免服药后血药浓度峰值,控制血药浓度为平稳曲线,提高药效持久性,同时,聚磷酸酯自身矿化后形成羟基磷灰石,在氨基酸的促进下,可以帮助骨组织修复与再生。本发明的技术方案是这样实现的:本发明提供一种抗肿瘤靶向药物缓释载体的制备方法,包括以下步骤:s1.聚磷酸酯-氨基酸共聚物的制备:将丙氨酸、酪氨酸、己内酰胺、去离子水和催化剂加入到反应器中,氮气保护下升温至第一温度反应,继续升温至第二温度反应,然后加入腺苷环磷酸酯反应0.5-1h,停止反应,待冷却至室温得到聚磷酸酯-氨基酸共聚物;s2.聚乳酸-羟基乙酸共聚物溶液的配置:称取聚乳酸-羟基乙酸共聚物,溶解于第一溶剂中,作为油相;s3.双亲化合物的制备:将聚磷酸酯-氨基酸共聚物溶于第二溶剂中,室温下反搅拌应2-4h,另将dmap溶于第二溶剂中,搅拌使其完全溶解,与步骤s2制得的聚乳酸-羟基乙酸共聚物溶液一起加入到反应体系中,继续在室温反应5-10h,反应结束采用冰无水乙醚沉淀,过滤,真空干燥,得到双亲化合物;s4.连接靶向分子的双亲化合物的制备:取双亲化合物溶于pbs缓冲溶液中,然后加入edc和nhs,超声15min后,搅拌条件下室温反应10-15h,再向该反应体系中加入端氨基叶酸,继续反应5-10h,加入去离子水搅拌均匀,冷却至室温,离心,取上清液,于去离子水中透析25-30h,每2-3小时换一次水以除去edc和nhs;然后冷冻干燥,得到连接靶向分子的双亲化合物;s5.纳米微球的制备:取步骤s4中连接靶向分子的双亲化合物溶解于第三溶剂中,配置成100-150mg/ml的油相,将抗肿瘤药物加入上述油相中,并加入正硅酸乙酯,搅拌至分散均匀,滴加去离子水,直到去离子水的体积为第三溶剂的5-10倍,继续加热至40-50℃,搅拌3-5h直至第三溶剂充分挥发,高速离心,冷冻干燥,得到纳米微球。作为本发明的进一步改进,步骤s1中所述第一温度为180-200℃,反应时间为0.5-2h;所述第二温度为200-220℃,反应时间为1-4h;所述催化剂选自二环己基碳二亚胺、二异丙基碳二亚胺、1-(3-二甲胺基丙基)-3-乙基碳二亚胺中的一种或几种混合;所述丙氨酸、酪氨酸、己内酰胺、腺苷环磷酸酯、催化剂的质量比为1:(0.5-1.2):(0.1-0.5):(0.5-0.7):(0.01-0.05)。作为本发明的进一步改进,步骤s2中所述第一溶剂为非极性溶剂,包括但不限于二氯甲烷、氯仿、乙酸乙酯、石油醚、乙酸甲酯、苯、甲苯、四氯化碳。作为本发明的进一步改进,步骤s3中所述第二溶剂选自二氯甲烷、氯仿、四氯化碳中的一种或几种混合,所述聚磷酸酯-氨基酸共聚物、聚乳酸-羟基乙酸共聚物溶液、dmap的质量比为1:5-10:(0.02-0.06)。作为本发明的进一步改进,步骤s4中所述pbs缓冲溶液的ph为7.2-7.5,所述双亲化合物、端氨基叶酸、edc和nhs的质量比为1:(0.2-0.5):(0.01-0.03):(0.01-0.02);所述离心转速为3000-5000r/min,离心时间为5-10min。作为本发明的进一步改进,步骤s5中所述第三溶剂为沸点不高于80℃的非极性溶剂,包括但不限于二氯甲烷、氯仿、四氯化碳、乙酸乙酯、乙酸甲酯;所述高速离心转速为10000-12000r/min,时间为2-4min;所述连接靶向分子的双亲化合物、抗肿瘤药物、正硅酸乙酯的质量比为1:(0.1-0.5):(0.5-1.2)。作为本发明的进一步改进,所述抗肿瘤药物为选自盐酸阿霉素、盐酸平阳霉素、盐酸表柔比星、盐酸吡柔比星、盐酸柔红霉素、高三尖杉酯碱、卡培他滨、长春新碱、羟基喜树碱、硫酸长春地辛、硫酸长春碱、重酒石酸长春瑞宾、紫杉醇、长春质碱、长春瑞宾碱、多烯紫杉醇、秋水仙碱、9-氨基喜树碱、7-乙基喜树碱、米托蒽醌或环磷酰胺中一种或多种。本发明进一步保护一种上述的制备方法制得的抗肿瘤靶向药物缓释载体。本发明进一步保护一种抗肿瘤靶向药物口服缓释制剂,含有上述的抗肿瘤靶向药物缓释载体。作为本发明的进一步改进,所述抗肿瘤靶向药物缓释载体的含量为5-10wt%。本发明通过验证,验证过程根据上述具体实施方式的步骤进行,用激光粒度仪测定粒径,用透射电镜观察纳米微球结构和粒径,用紫外分光光度计测定载药量和研究其释放特性。获得了较好的验证结果。其中纳米微球粒径在220nm左右,表面光滑,载药量为14.5-17.2%,包封率为70.2-77.7%,本发明的产品的一个较稳定的一个范围值为载药量为15.2-16.7%,包封率为73.4-75.6%。释药能持续缓慢释放250小时以上。本发明具有如下有益效果:本发明采用聚磷酸酯-氨基酸共聚物修饰的聚乳酸-羟基乙酸共聚物是具有疏水端和亲水端的两性化合物,抗肿瘤药物接枝在疏水部分,叶酸靶向分子接枝在亲水部分,形成带有靶向分子和抗肿瘤药物的长链分子,自组装作用下形成载药纳米微球或者纳米胶束,在溶胶凝胶法水解时,正硅酸乙酯水解形成的si-o-si时形成氢键,并在亲疏水片段之间形成一层硅壳。聚乳酸-羟基乙酸共聚物段是疏水部分,具备良好的生物可降解性、生物相容性和适当的降解速率的特点,可以通过调节乳酸和羟基乙酸的比例来控制其被生物降解的快慢,且聚乳酸-羟基乙酸共聚物应用没有安全隐患,因为其降解后产生的物质是人体本身就存有的乳酸、羟基乙酸;聚磷酸酯-氨基酸共聚物是亲水部分,聚磷酸酯本身不可降解,在一定程度上经生物基聚酰胺修饰的载体材料会减少聚乳酸-羟基乙酸共聚物局部产酸量,同时,聚磷酸酯自身矿化后形成羟基磷灰石,在氨基酸的促进下,可以帮助骨组织修复与再生;本发明以腺苷环磷酸酯为原料与丙氨酸、酪氨酸、己内酰胺发生反应,引入了磷酸酯基团、酰胺基团和丙氨酸、酪氨酸结构,肿瘤细胞中含有相比正常细胞含有较高浓度的磷酸酯酶和酰胺酶,以及酪氨酸蛋白酶的活性和含量比正常的细胞高,其均可用于肿瘤细胞定位,与叶酸进行协同定位,大大提高了药物的选择性和定位的准确性,经酶水解后释放出抗肿瘤药物,高效杀灭癌细胞,提高药效,缓释制剂可以有效避免服药后血药浓度峰值,控制血药浓度为平稳曲线,提高药效持久性。附图说明为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动性的前提下,还可以根据这些附图获得其他的附图。图1为本发明实施例1中步骤s1中反应路线图;图2为本发明实施例1中步骤s3中反应路线图;图3为本发明实施例1中步骤s4中反应路线图;图4为本发明实施例1中步骤s5中反应路线图;图5为本发明实施例3中制得的纳米微球的sem图;图6为本发明实施例6中口服缓释制剂的药物释放曲线图。具体实施方式下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。实施例1抗肿瘤靶向药物缓释载体的制备方法s1.聚磷酸酯-氨基酸共聚物的制备:将1g丙氨酸、0.5g酪氨酸、0.1g己内酰胺、20ml去离子水和0.01g1-(3-二甲胺基丙基)-3-乙基碳二亚胺加入到反应器中,氮气保护下升温至180℃,反应时间为0.5h,继续升温至200℃,反应时间为1h,然后加入0.5g腺苷环磷酸酯反应0.5h,停止反应,待冷却至室温得到聚磷酸酯-氨基酸共聚物;反应路线图如图1所示;s2.聚乳酸-羟基乙酸共聚物溶液的配置:称取1g聚乳酸-羟基乙酸共聚物,溶解于10ml四氯化碳中,作为油相;s3.双亲化合物的制备:将1g聚磷酸酯-氨基酸共聚物溶于10ml二氯甲烷中,室温下反搅拌应2h,另将0.01gdmap溶于10ml二氯甲烷中,搅拌使其完全溶解,与5g步骤s2制得的聚乳酸-羟基乙酸共聚物溶液一起加入到反应体系中,继续在室温反应5h,反应结束采用冰无水乙醚沉淀,过滤,真空干燥,得到双亲化合物;反应路线图如图2所示;s4.连接靶向分子的双亲化合物的制备:取1g双亲化合物溶于pbs缓冲溶液(ph=7.2)中,然后加入0.01gedc和0.01gnhs,超声15min后,搅拌条件下室温反应10h,再向该反应体系中加入0.2g端氨基叶酸,继续反应5h,加入去离子水搅拌均匀,冷却至室温,离心,离心转速为3000r/min,离心时间为5min,取上清液,于去离子水中透析25h,每2小时换一次水以除去edc和nhs;然后冷冻干燥,得到连接靶向分子的双亲化合物;反应路线图如图3所示;s5.纳米微球的制备:取1g步骤s4中连接靶向分子的双亲化合物溶解于二氯甲烷中,配置成100mg/ml的油相,将0.1g盐酸柔红霉素加入上述油相中,并加入0.5g正硅酸乙酯,300/min搅拌至分散均匀,滴加去离子水,直到去离子水的体积为二氯甲烷的5倍,继续加热至40℃,搅拌3h直至二氯甲烷充分挥发,高速离心,离心转速为10000r/min,时间为2min,冷冻干燥,得到纳米微球;反应路线图如图4所示。实施例2抗肿瘤靶向药物缓释载体的制备方法s1.聚磷酸酯-氨基酸共聚物(ppe-aa)的制备:将1g丙氨酸、1.2g酪氨酸、0.5g己内酰胺、20ml去离子水和0.05g二环己基碳二亚胺加入到反应器中,氮气保护下升温至200℃,反应时间为2h,继续升温至220℃,反应时间为4h,然后加入0.7g腺苷环磷酸酯反应1h,停止反应,待冷却至室温得到聚磷酸酯-氨基酸共聚物;s2.聚乳酸-羟基乙酸共聚物(plga)溶液的配置:称取1g聚乳酸-羟基乙酸共聚物,溶解于10ml二氯甲烷中,作为油相;s3.双亲化合物(ppe-aa-plga)的制备:将1g聚磷酸酯-氨基酸共聚物溶于10ml二氯甲烷中,室温下反搅拌应4h,另将0.04gdmap溶于10ml二氯甲烷中,搅拌使其完全溶解,与10g步骤s2制得的聚乳酸-羟基乙酸共聚物溶液一起加入到反应体系中,继续在室温反应10h,反应结束采用冰无水乙醚沉淀,过滤,真空干燥,得到双亲化合物;s4.连接靶向分子的双亲化合物(fa-ppe-aa-plga)的制备:取1g双亲化合物溶于pbs缓冲溶液(ph=7.5)中,然后加入0.03gedc和0.02gnhs,超声15min后,搅拌条件下室温反应15h,再向该反应体系中加入0.5g端氨基叶酸,继续反应10h,加入去离子水搅拌均匀,冷却至室温,离心,离心转速为5000r/min,离心时间为10min,取上清液,于去离子水中透析30h,每3小时换一次水以除去edc和nhs;然后冷冻干燥,得到连接靶向分子的双亲化合物;s5.纳米微球(fa-ppe-aa-plga-cap)的制备:取1g步骤s4中连接靶向分子的双亲化合物溶解于乙酸乙酯中,配置成150mg/ml的油相,将0.5g卡培他滨加入上述油相中,并加入1.2g正硅酸乙酯,300/min搅拌至分散均匀,滴加去离子水,直到去离子水的体积为乙酸乙酯的10倍,继续加热至50℃,搅拌5h直至乙酸乙酯充分挥发,高速离心,离心转速为12000r/min,时间为4min,冷冻干燥,得到纳米微球。图5为制得的纳米微球的sem图,有图可知,微球的粒径在220nm左右。实施例3本实施例与实施例2的不同之处在于:将实施例1中的步骤s5中去离子水的加入体积为乙酸乙酯的50倍。实施例4本实施例与实施例2不同之处在于:将实施例2中的步骤s1中升温温度调整:第一次升温温度为160℃,第二次升温温度为180℃。实施例5本实施例与实施例2不同之处在于:将实施例2中的步骤s5中取1g步骤s4中连接靶向分子的双亲化合物溶解于乙酸乙酯中,配置浓度调整为50mg/ml的油相。实施例6本实施例与实施例2的不同之处在于:将实施例2中的步骤s5中搅拌转速调整为500r/min。实施例7一种抗肿瘤靶向药物口服缓释制剂为口服液剂型,含有实施例2制得的抗肿瘤靶向药物缓释载体的含量为10wt%,余量为生理盐水。将本实施例制剂给药后释放结果如图6所示,释放速率由快变慢,但不是突释效果,能持续给药长达11d,11d后药物释放率达到90%,而后再缓慢释放,说明实施例2制备的抗肿瘤靶向药物缓释载体是种理想的药物缓释载体。实施例8一种抗肿瘤靶向药物口服缓释制剂为片剂剂型,含有实施例2制得的抗肿瘤靶向药物缓释载体的含量为5wt%,硬脂酸镁2.5wt%,微晶纤维素1.2wt%,余量为麦芽糊精。测试例1用激光粒度仪和紫外分光光度计对所得到的纳米微球进行测试表征实施例1-6制得的纳米微球的粒径和粒径分布,载药量和包封率如表1所示。表1组别粒径(nm)粒径分布指数载药量(%)包封率(%)实施例1222±0.330.32516.274.7实施例2219±0.450.31716.675.2实施例3165±0.520.4577.245.3实施例4155±0.110.66211.262.5实施例5242±0.250.52412.564.1实施例6265±0.170.71210.460.2实施例3中纳米微球的粒径较实施例2中小,原因是水相体积的增大,减小了油相的粘度,有利于油相在水相中的分散形成乳滴,使得最终形成的微球粒径减小。实施例4中纳米微球的粒径较实施例2中小,原因是步骤s1中聚磷酸酯-氨基酸共聚物的反应温度较低,形成的共聚物未能充分反应,使得共聚物含量较少,制得的微球粒径较小。实施例5中纳米微球的粒径较实施例2中大,原因是连接靶向分子的双亲化合物配置成的油相浓度较低,油相的粘度较大,在水相中形成较大的乳滴,使得最终的产品粒径增大。实施例6中随着搅拌速度的增加,纳米微球的粒径逐渐减小,同时粒径分布也在一定程度上变窄,而且微球的包封率和载药量也下降。原因是随着搅拌速度的增大,剪切力也增大,导致油相液滴在水相中的分散程度提高,使得nps更易分散为较小的乳滴,但因为搅拌剧烈,导致了药物的泄露,故包封率和载药量显著减小。粒径在250nm以内的纳米制剂由于肿瘤组织的epr效应可被动靶向于肿瘤组织,而减少了对正常组织的杀伤作用。如聚合物载药胶束,脂质体纳米颗粒等在人体的内循环中能够很顺利的进出肿瘤组织细胞,进而在肿瘤组织累积必须控制在较小直径。而包封率和载药量是评价纳米制剂质量的重要指标,包封率越大,药物损失越少,而载药量越大,越容易满足临床用药的需求。与现有技术相比,本发明采用聚磷酸酯-氨基酸共聚物修饰的聚乳酸-羟基乙酸共聚物是具有疏水端和亲水端的两性化合物,抗肿瘤药物接枝在疏水部分,叶酸靶向分子接枝在亲水部分,形成带有靶向分子和抗肿瘤药物的长链分子,自组装作用下形成载药纳米微球或者纳米胶束,在溶胶凝胶法水解时,正硅酸乙酯水解形成的si-o-si时形成氢键,并在亲疏水片段之间形成一层硅壳。聚乳酸-羟基乙酸共聚物段是疏水部分,具备良好的生物可降解性、生物相容性和适当的降解速率的特点,可以通过调节乳酸和羟基乙酸的比例来控制其被生物降解的快慢,且聚乳酸-羟基乙酸共聚物应用没有安全隐患,因为其降解后产生的物质是人体本身就存有的乳酸、羟基乙酸;聚磷酸酯-氨基酸共聚物是亲水部分,聚磷酸酯本身不可降解,在一定程度上经生物基聚酰胺修饰的载体材料会减少聚乳酸-羟基乙酸共聚物局部产酸量,同时,聚磷酸酯自身矿化后形成羟基磷灰石,在氨基酸的促进下,可以帮助骨组织修复与再生;本发明以腺苷环磷酸酯为原料与丙氨酸、酪氨酸、己内酰胺发生反应,引入了磷酸酯基团、酰胺基团和丙氨酸、酪氨酸结构,肿瘤细胞中含有相比正常细胞含有较高浓度的磷酸酯酶和酰胺酶,以及酪氨酸蛋白酶的活性和含量比正常的细胞高,其均可用于肿瘤细胞定位,与叶酸进行协同定位,大大提高了药物的选择性和定位的准确性,经酶水解后释放出抗肿瘤药物,高效杀灭癌细胞,提高药效,缓释制剂可以有效避免服药后血药浓度峰值,控制血药浓度为平稳曲线,提高药效持久性。以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。当前第1页1 2 3
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询




tips