
 商标分类
商标分类  商标转让
商标转让 
一种芳胺类化合物及其制备的液晶取向剂、液晶取向膜和液晶显示元件的制作方法
 2021-02-02 22:02:02|
2021-02-02 22:02:02| 479|
479| 起点商标网
起点商标网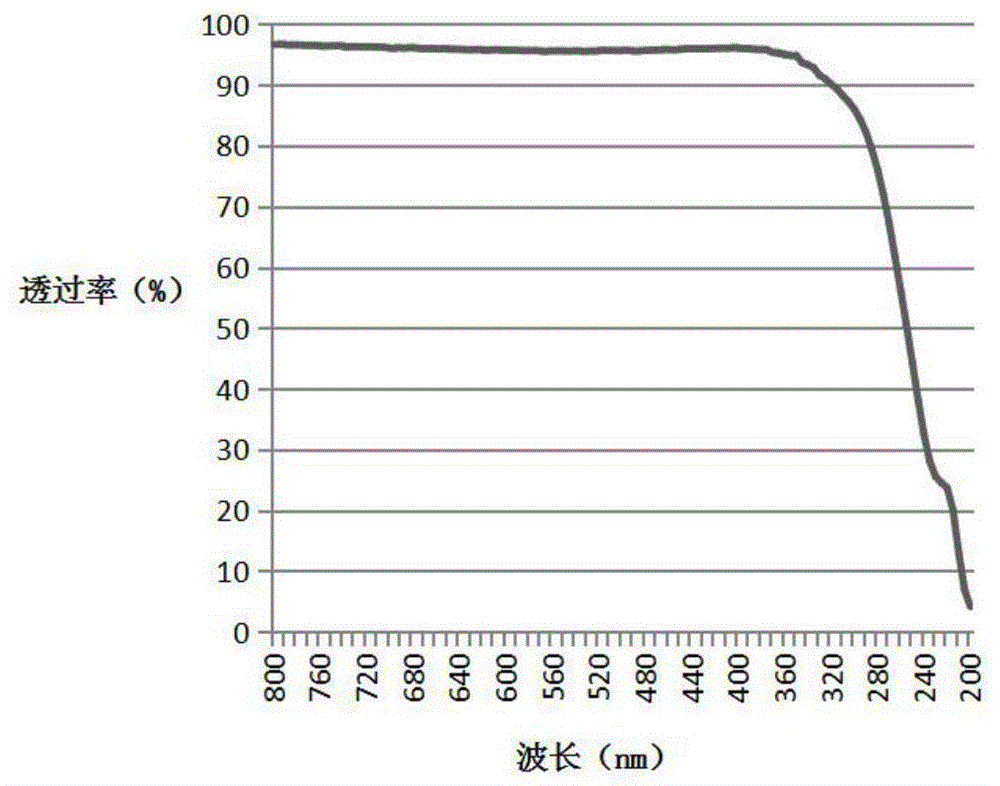
本发明涉及液晶显示技术领域,尤其是涉及一种芳胺类化合物及其制备的液晶取向剂、液晶取向膜以及液晶显示元件。
背景技术:
液晶显示元件根据电极结构及所用的液晶分子的物性分类成各种模式。目前已知的液晶显示装置分类如下,扭转向列(twistednematic,tn)型或超扭转向列(supertwistednematic,stn)型、边缘场切换(fringefieldswitching,ffs)型垂直取向(verticalalignment,va)型、共面切换(in-planeswitching,ips)型等各种液晶显示元件。液晶取向膜在液晶显示元件中使液晶分子发生取向,聚酰亚胺凭借其优良的耐热性、抗腐蚀性能、机械强度、与液晶的亲和性能优异,成为作为液晶取向膜的首选材料。
液晶显示元件的技术的发展不仅是通过原件结构和驱动方式的改良来实现,而且还通过元件中所使用的组成构件来实现。液晶显示元件中液晶取向膜是关系到显示品质的重要材料之一,伴随液晶显示元件的高品质化,提高液晶取向膜的性能变得至观重要。液晶显示元件中,若使用透过率低的液晶取向膜,则会产生液晶显示元件的亮度降低的问题。同时对比度也是液晶液晶显示元件的一个重要参数,在合理的亮度值下,对比度越高,其所能现实的色彩层次越丰富,视觉感更强,图像越清晰醒目;而对比度小,则让整个画面表现为灰暗。
技术实现要素:
针对现有技术存在的问题,本发明申请人提供了一种芳胺类化合物及其制备的液晶取向剂、液晶取向膜和液晶显示元件。本发明的液晶取向剂所制得的液晶取向膜具有成膜透过率高和所制得的液晶显示元件具有对比度高的优点。
本发明解决上述技术问题的技术方案如下:
一种芳胺类化合物,所述芳胺化合物的结构如通式(1)所示:
通式(1)中,r1、r2分别独立的表示为h原子或通式(2)所示结构,且r1、r2不同时为h原子;x1、x2分别独立地表示为氢原子、卤素原子、c1-5烷基中的一种;
通式(2)中,r3表示为c1-5的亚烷基;r4、r5分别独立的表示为h原子、c1-5烷基、叔丁氧羰基或叔丁氧羰基取代的烷基。
优选方案,所述芳胺类化合物的具体结构如式1-1至式1-5所示:
一种所述芳胺类化合物制备的液晶取向剂,所述液晶取向剂包括四羧酸二酐组分a与胺类组分b反应得到的聚合物a;所述胺类组分含有所述的芳胺类化合物b-1。
所述四羧酸二酐组分a至少包括1,2,3,4-环丁烷四羧酸二酐、1,3-二甲基-1,2,3,4-环丁烷四羧酸二酐中的一种。
所述四羧酸二酐组份a还包括3,3’,4,4’-联苯砜四羧酸二酐、均苯四羧酸二酐、1,2,4,5-环己烷四羧酸二酐、3,3’,4,4’-联苯四羧酸二酐、1,2,3,4-环戊烷四羧酸二酐、2,3,5-三羧基环戊基乙酸二酐中的一种或多种。
所述胺类组分b包括其它芳胺化合物b-2,所述其它芳胺化合物b-2为对苯二胺、间苯二胺、2,2-双(4-氨基苯基)六氟丙烷、4-(4-庚基环己基)苯基-3,5-二氨基苯甲酸酯、2,2’-二甲基-4,4’-二氨基联苯、4,4’-二氨基二苯醚、1,4-二(4-氨基苯氧基)苯、4,4’-二氨基二苯甲酮、1,2-双(4-氨基苯氧基)乙烷、1,3-双(4-氨基苯氧基)丙烷、1,4-双(4-氨基苯氧基)丁烷、1,5-双(4-氨基苯氧基)戊烷、1,6-双(4-氨基苯氧基)己烷、n,n’-二(4-氨基苯基)哌嗪、1,5-二氨基萘、1,8-二氨基萘、对氨基苯乙胺、4,4’-二氨基二苯甲烷、4,4’-二氨基二苯乙烷、4,4’-二氨基苯甲酰胺、2,2-双[4-(4-氨基苯氧基)苯基]六氟丙烷、2,2-双[4-(4-氨基苯氧基)苯基]丙烷、2,4-二氨基十二烷氧基苯、2,4-二氨基十八烷氧基苯、1-(4-(4-戊基环己基环己基)苯氧基)-2,4-二氨基苯、1-(4-(4-庚基环己基)苯氧基)-2,4-二氨基苯、3,5-二氨基苯甲酸中的一种或多种。
所述液晶取向剂还包括溶剂组分,所述溶剂组份为n-甲基-2-吡咯烷酮、γ-丁内酯、n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、乙二醇单乙醚、乙二醇单丁醚、乙二醇单甲醚、乙二醇甲乙醚、乙二醇二甲醚、二甘醇单甲醚乙酯中的一种或多种。
一种液晶取向膜,包括所述的液晶取向剂。
一种液晶显示元件,包括所述的液晶取向膜。
四羧酸二酐组分a与胺类组分b反应得到的聚合物a,所述聚合物a为聚酰胺酸,其制备方法,包括如下步骤:将包含四羧酸二酐组分a和芳胺组分b在有机溶剂的存在下,在-20℃~150℃、优选0℃~100℃下反应30分钟-48小时、优选1-24小时来合成得到聚酰胺酸溶液。用于聚合反应的溶剂与液晶取向剂中的溶剂可以相同或不同,且用于聚合反应的溶剂并无特别的限制,只要能溶解单体和聚合物即可。聚合反应的溶剂包括但不限于n-甲基-2-吡咯烷酮、n,n-二甲基乙酰胺、n,n-二甲基甲酰胺、γ-丁内酯,也可以是一种或两种以上混合使用。其中,四羧酸二酐组分a和芳胺组分b混合得到的反应混合物和所述溶剂组成的反应液中,反应混合物占反应液重量比为1-50%,更优选为10-25%。
所得到聚酰胺酸溶液可以在减压下蒸掉溶剂得到聚酰胺酸固体,或者在充分搅拌下将反应体系倾倒入大量的溶剂中,通过进行几次析出、用溶剂洗涤后,常温或加热烘干,得到聚酰胺酸固体粉末。析出所用的溶剂并无特别的限定,只要所用溶剂可以将聚酰胺酸析出即可,可列举为甲醇、乙醇、水、丙醇、异丙醇、丁醇、丁基溶纤剂、甲苯、乙酸乙酯、乙羧以及含卤素溶剂中的一种或几种的混合液。
进一步,将聚合反应制得的聚酰胺酸固体粉末溶于溶剂中,制得液晶取向剂,其中聚酰胺酸固体所占的重量比为1-40%,更优选为2-20%。
在液晶取向剂中聚酰胺酸固体所占重量比小于2%的情况下,涂膜的膜厚变得过小而难以获得良好的液晶取向膜,另一方面,在液晶取向剂中聚酰胺酸固体所占重量比超过20%的情况下,涂膜的膜厚变得过大而难以获得良好的液晶取向膜,因此更优选为2%-20%。
进一步,所述四羧酸二酐组份a和芳胺组分b的摩尔比为100:10-200,更优选为100:100-120。
进一步,所述芳胺组分b中,芳胺化合物b-1所占的摩尔百分比为0.1-100%,更优选为5-70%。
进一步,本发明的液晶取向剂中还可包括降低溶液表面张力以及增加取向剂流平效果作用的溶剂,所述溶剂包括甲醇、乙醇、异丙醇、环己醇、乙二醇、四氢呋喃、二氯甲烷、氯苯、1,2-二氯乙烷、丙酮、甲乙酮、甲基异丁酮、环丁酮、乙酸甲酯、乙酸乙酯、乙醚、乙二醇单甲醚、乙二醇单乙醚、乙二醇单丁醚、乙二醇甲乙醚、乙二醇二甲醚中的一种或多种,所占溶剂组分总重量的0-70%,优选地,所占溶剂总重量的10-60%。
本发明进一步提供了聚酰亚胺的制备方法,可以采用但不限于以下两种亚胺化方法,此两种亚胺化方法是指热亚胺化法或化学亚胺化法。
热亚胺化法是指将聚酰亚胺固体直接加热脱水成环,加热温度优选为150-300℃。
化学酰亚胺化是在较低的温度下进行酰亚胺化反应,在酰亚胺化的过程中不易发生聚合物的水解而分子量下降。
化学亚胺化法包括如下步骤:聚酰胺酸在脱水剂和催化剂存在下,通过较低的温度脱水关环制备聚酰亚胺。
亚胺化反应的溶剂可以与液晶取向剂中的溶剂相同。
其中,所述聚酰胺酸和所述亚胺化反应溶剂的重量比为1:2-25;聚酰胺酸的亚胺化率为10-100%;亚胺化反应的温度为0-110℃,更优选为30-80℃;反应时间为1-100小时,更优选为2-10小时;脱水剂可以选择一个酸酐类化合物,比如醋酸酐、偏苯三酸酐、均苯四甲酸酐、丙酸酐或三氟醋酸酐;聚酰胺酸中所用原材料四酸二酐和脱水剂的摩尔比优选为1:0.1-10,更优选为1:1-5;催化剂可选自吡啶、4-甲基吡啶、三甲胺、三乙胺、三丁胺或三辛胺;所述脱水剂和所述催化剂的摩尔比为1:0.1-5,更优选为1:2-4。
在聚酰胺酸的酰亚胺化反应后的溶液中残留所添加的催化剂等,可以通过将酰亚胺化聚合物回收、有机溶剂再溶解,形成本发明的液晶取向剂。
将上所述得到的聚酰亚胺的溶液通过充分搅拌的同时注入到溶剂中,可以使聚合物析出。进行几次析出后、进行烘干后可以得到经过纯化的聚酰胺酸的粉末。
所述析出溶剂没有特别限定,可列举出甲醇、丙酮、己烷、丁基溶纤剂、庚烷、甲乙酮、甲基异丁基酮、乙醇、甲苯、苯等。
本发明进一步提供了液晶取向剂的制备方法,
本发明的液晶取向剂含有聚合物a。本发明的液晶取向剂中,除了聚合物a之外,还可以含有其他聚合物,例如:聚酰胺酸、聚酰亚胺、聚酰胺酸酯、聚酯、聚酰胺等。
进一步,本发明的液晶取向剂中还可包括分子量调节剂,通过在聚合物的合成过程中加入分子量调节剂来调节聚合物的分子量,保证后续涂覆工艺的可行性。所述分子量调节剂包括正庚胺、异氰酸苯酯、苯胺、正丁胺、正戊胺、正己胺、异氰酸萘酯、马来酸酐、邻苯二甲酸酐、邻环己二甲酸酐、琥珀酸酐中的一种或多种,分子量调节剂与四羧酸二酐组分a的摩尔比为0.001-15:100。优选地,分子量调节剂与四羧酸二酐组分a的摩尔比为0.4-6:100。
进一步,本发明的液晶取向剂中还可包括添加剂,添加剂的作用是增加液晶取向膜的稳定性或者提高液晶取向膜与基板之间的附着力,液晶取向剂可通过将聚合物和添加剂在溶剂中于10-100℃在搅拌下混合制得,更优选为25-75℃。
所述添加剂包括环氧类添加剂和/或具有官能性基团的硅烷类化合物添加剂,环氧类添加剂的加入量为液晶取相剂中聚酰胺酸或聚酰亚胺总重量的0.01-15%,优选地,环氧类添加剂的加入量为聚合物总重量的0.5-10%,具有官能性基团的硅烷类化合物添加剂的加入量为聚合物总重量的0.01-10%,优选地,具有官能性基团的硅烷类化合物添加剂的加入量为聚合物总重量的0.5-5%;
环氧类添加剂为聚丙二醇二环氧丙基醚、聚乙二醇二环氧丙基醚、丙二醇二环氧丙基醚、丙三醇二环氧丙基醚、1,6-已二醇二环氧丙基醚、乙二醇二环氧丙基醚、n,n,n’,n’-四环氧丙基-4,4’-二氨基二苯甲烷或3-(n,n-二环氧丙基)氨基丙基三甲氧基硅烷中的一种或多种;
具有官能性基团的硅烷类化合物添加剂为3-氨基丙基三乙氧基硅烷、2-氨基丙基三甲氧基硅烷、3-氨基丙基三乙氧基硅烷、n-(2-氨基乙基)-3-氨基丙基甲二甲氧基硅烷、n-(2-氨基乙基)-3-氨基丙基三甲氧基硅烷、3-氨基丙基三甲氧基硅烷、n-苯基-3-氨基丙基三甲氧基硅烷或n-双(氧化乙烯)-3-氨基丙基三乙氧基硅烷中的一种或多种。
本发明还提供了一种前述液晶取向剂在液晶取向膜的应用,利用前述液晶取向剂制成液晶取向膜。
本发明的液晶取向膜含有本发明的液晶取向剂,而液晶取向剂制备所用芳胺单体中含有特定结构的芳胺化合物b-1,制得的液晶取向剂颜色浅,同时b-1单体中的极性基团可以提高其对表面液晶分子的范德华力,因此提高了其对液晶分子的锚定能力,液晶显示元件上表现出对比度高的特点。因此本发明的液晶取向剂所制得的液晶取向膜具有成膜透过率高和所制得的液晶显示元件具有对比度高的优点。体现在实施例中,利用该取向剂制备的液晶取向膜透过率评估:95%以上判断为最优;对比度评估:利用该取向剂制备的液晶显示元件对比度结果为大于等于1800时,评估为对比度优秀。
液晶取向膜是将液晶取向剂涂布到基板上,固化后获得的膜。液晶取向膜的取向方法并无特别的限制,可以采用但不限于光取向方法或摩擦取向的方法。其中,光取向是指用偏振光对膜表面进行曝光处理,形成光取向的液晶取向膜;摩擦取向是指用尼龙、人造丝、棉类或其他纤维所做成的布料缠绕在滚筒上,并以一定方向摩擦取向膜,形成摩擦取向的取向膜。
本发明还提供了一种前述液晶取向剂在液晶显示元件的应用,利用前述液晶取向剂制成液晶显示元件。
上述液晶显示元件的制备方法,包括如下步骤:准备两片基板,每片基板上均涂有一层由本发明的液晶取向剂所制得的液晶取向膜,并于两层基板中间充满液晶以制得一个液晶盒。
用本发明中的液晶取向剂制得的液晶显示元件适合各种类型的液晶显示元件,可以是扭转向列(twistednematic,tn)型或超扭转向列(supertwistednematic,stn)型、垂直取向(verticalalignment,va)型、共面切换(in-planeswitching,ips)型、边缘场切换(fringefieldswitching,ffs)型等。在上述液晶显示元件中,优选ips和ffs型液晶显示元件。
本发明有益的技术效果在:
本发明的液晶取向剂的液晶取向膜具有透过率高的特性,对于有益效果产生的机理尚不明确,可以认为是由于所用的特定的芳胺化合物b-1单体所引起的可见光区域的光的吸收少,因此取向剂制得的液晶取向膜具有透过率高的特性。
本发明的液晶取向剂的液晶取向膜具有对比度高的特性,对于有益效果产生的机理尚不明确,可以认为是由于所用的特定的芳胺化合物b-1单体中的极性基团可以提高其对表面液晶分子的范德华力,因此提高了其对液晶分子的锚定能力,液晶显示元件上表现出对比度高的特点。
本发明方法简单,市场前景广阔,适合于规模化应用推广。
附图说明
图1是实施例1中所获得的液晶取向膜的uv-vis光谱。
图2是比较例1中所获得的液晶取向膜的uv-vis光谱。
具体实施方式
在下面具体例中,仅以ips和ffs型的液晶显示元件对该液晶取向剂进行说明,但本发明并不限于此。
(一)化合物的合成例
芳胺化合物的合成例
化合物合成例1
结构式(1-1)所代表的化合物可根据下面的合成路线1来合成:
(1)化合物1-1-a的合成
于1l的三口圆底烧瓶中投入2,4-二氨基碘苯(23.40g,100毫摩尔)、et3n(50.60g,500毫摩尔)和200g甲苯,将体系升温到60℃,然后缓慢向体系内滴加3-硝基-4-甲基苯甲酰氯(41.91g,210毫摩尔)和200g甲苯的混合液,0.5小时滴加完成,然后将体系升温到80℃搅拌8至10小时,tlc跟踪,直至原料2,4-二氨基碘苯无剩余。然后将反应体系降至室温,用500ml*3的去离子水,水洗三次,上层有机相脱干得到淡黄色固体,加入500ml甲醇/水的混合液打浆,此悬浊液经过滤烘干以75%收率得到化合物1-1-a。
(2)化合物1-1-b的合成
于1l的三口圆底烧瓶中投入1-1-a(56.03g,100毫摩尔)、pd(pph3)4(2.31g,2毫摩尔)、cui(0.38g,2毫摩尔)、et3n(50.60g,500毫摩尔)和dmf溶剂200ml,在20-40℃搅拌30min,而后向体系内滴加炔戊基胺(9.14g,110毫摩尔)和100ml溶剂dmf的混合液,1-2小时滴加完毕,于20-40℃保温反应4-10小时。
反应结束后,向体系中加入500ml1,2-二氯乙烷和500ml浓度为1mol/l的盐酸溶液,有机相用500ml*3的去离子水,水洗三次,机相脱干得到淡黄色固体,以63%收率得到化合物1-1-b。
(3)化合物1-1的合成
于1l的高压釜中投入获得的化合物1-1-b(25.78g,50毫摩尔)、5%的钯碳(2.86g,含水,固含量为45%)和600g四氢呋喃,将高压釜密封,用氢气置换3-5次后,氢气加压至0.5-1.0mpa,搅拌下于45-55℃反应。反应结束后,用孔径为0.2μm的滤膜将催化剂滤出,滤液脱溶剂,所得固体加入60g乙醇并搅拌30min,经过抽滤和烘干,将以92%的收率得到一种类白色的固体化合物1-1。
该化合物1-1的高分辩率质谱,esi源,正离子模式,分子式c27h33n5o2,理论值459.26,测试值459.48。元素分析(c27h33n5o2),理论值c:70.59,h:7.24,n:15.24,o:6.96,实测值c:70.61,h:7.23,n:15.25,o:6.94。元素分析测试仪品牌:美国加联,型号:ce-440。
化合物合成例2
结构式(1-2)所代表的化合物可根据下面的合成路线2来合成:
(1)化合物1-2-a的合成
于1l的三口圆底烧瓶中投入1,4-二氨基-2,5-二碘苯(35.99g,100毫摩尔)、et3n(50.60g,500毫摩尔)和200g甲苯,将体系升温到60℃,然后缓慢向体系内滴加2-硝基-5-氟苯甲酰氯(42.75g,210毫摩尔)和200g甲苯的混合液,约0.5小时滴加完成,然后将体系升温到80℃搅拌8至10小时,tlc跟踪,直至原料1,4-二氨基-2,5-二碘苯无剩余。然后将反应体系降至室温,用500ml*3的去离子水,水洗三次,上层有机相脱干得到淡黄色固体,加入500ml甲醇/水的混合液打浆,此悬浊液经过滤烘干以76%收率得到化合物1-2-a。
(2)化合物1-2-b的合成
于1l的三口圆底烧瓶中投入1-2-a(64.91g,100毫摩尔)、pd(pph3)4(2.31g,2毫摩尔)、cui(0.38g,2毫摩尔)、et3n(50.60g,500毫摩尔)和dmf溶剂200ml,在20-40℃搅拌30min,而后向体系内滴加炔丁基胺(7.60g,110毫摩尔)和100ml溶剂dmf的混合液,1-2小时滴加完毕,升温到20-40℃保温反应4-10小时。
反应结束后,向体系中加入500ml1,2-二氯乙烷和500ml浓度为1mol/l的盐酸溶液,有机相用500ml*3的去离子水,水洗三次,机相脱干得到淡黄色固体,以65%收率得到化合物1-1-b。
(3)化合物1-2-c的合成
于1l的三口圆底烧瓶中投入1-2-b(57.65g,100毫摩尔)、碘乙烷(32.75g,210毫摩尔)、叔丁醇钾(23.56g,210毫摩尔)、醋酸钯(0.224g,1毫摩尔)、xphos(0.953g,2毫摩尔)和300g甲苯,浴温升至110℃搅拌回流3小时,tlc跟踪,直至原料1-2-b无剩余,把反应液转移分液漏斗中水洗至中性,脱除溶剂甲苯,滤液浓缩至重量为100g,加入220g乙醇,有黄色固体析出,抽滤,烘干滤饼得黄色晶体49.98g,,此反应收率为79%。
(4)化合物1-2的合成
于1l的高压釜中投入获得的化合物1-2-c(31.63g,50毫摩尔)、5%的钯碳(3.51g,含水,固含量为45%)和600g四氢呋喃,将高压釜密封,用氢气置换3-5次后,氢气加压至0.5-1.0mpa,搅拌下于45-55℃反应。反应结束后,用孔径为0.2μm的滤膜将催化剂滤出,滤液脱溶剂,所得固体加入60g乙醇并搅拌30min,经过抽滤和烘干,将以90%的收率得到一种类白色的固体化合物1-2。
该化合物1-2的高分辩率质谱,esi源,正离子模式,分子式c32h42f2n6o2,理论值580.33,测试值580.58。元素分析(c32h42f2n6o2),理论值c:66.18,h:7.29,f:6.54,n:14.47,o:5.51,实测值c:66.16,h:7.30,f:6.55,n:14.46,o:5.52。元素分析测试仪品牌:美国加联,型号:ce-440。
化合物合成例3
结构式(1-3)所代表的化合物可根据下面的合成路线3来合成:
(1)化合物1-3-a的合成
于1l的三口圆底烧瓶中投入2,4-二氨基碘苯(23.40g,100毫摩尔)、et3n(50.60g,500毫摩尔)和200g甲苯,将体系升温到60℃,然后缓慢向体系内滴加对硝基苯甲酰氯(38.97g,210毫摩尔)和200g甲苯的混合液,约0.5小时滴加完成,然后将体系升温到80℃搅拌8至10小时,tlc跟踪,直至原料2,4-二氨基碘苯无剩余。然后将反应体系降至室温,用500ml*3的去离子水,水洗三次,上层有机相脱干得到淡黄色固体,加入500ml甲醇/水的混合液打浆,此悬浊液经过滤烘干以73%收率得到化合物1-3-a。
(2)化合物1-3-b的合成
于1l的三口圆底烧瓶中投入1-3-a(53.23g,100毫摩尔)、pd(pph3)4(2.31g,2毫摩尔)、cui(0.38g,2毫摩尔)、et3n(50.60g,500毫摩尔)和dmf溶剂200ml,在20-40℃搅拌30min,而后向体系内滴加炔丙基胺(6.06g,110毫摩尔)和100ml溶剂dmf的混合液,1-2小时滴加完毕,于20-40℃保温反应4-10小时。
反应结束后,向体系中加入500ml1,2-二氯乙烷和500ml浓度为1mol/l的盐酸溶液,有机相用500ml*3的去离子水,水洗三次,机相脱干得到淡黄色固体,收率68%。
(3)化合物1-3-c的合成
于1l的三口圆底烧瓶中投入1-3-b(45.94g,100毫摩尔)和300g二氯乙烷,降温至0-10℃,滴加入二碳酸二叔丁酯(21.83g,100毫摩尔)的200g二氯乙烷溶液,滴加用时0.5-1小时。滴加完毕后室温搅拌24小时。反应结束后,反应液用500ml去离子水水洗,脱除二氯甲烷,得到49.8g淡黄色固体,收率为89%。
(4)化合物1-3的合成
于1l的高压釜中投入获得的化合物1-3-c(27.98g,50毫摩尔)、5%的钯碳(3.11g,含水,固含量为45%)和600g四氢呋喃,将高压釜密封,用氢气置换3-5次后,氢气加压至0.5-1.0mpa,搅拌下于45-55℃反应。反应结束后,用孔径为0.2μm的滤膜将催化剂滤出,滤液脱溶剂,所得固体加入60g乙醇并搅拌30min,经过抽滤和烘干,将以92%的收率得到一种类白色的固体化合物1-3。
该化合物1-3的高分辩率质谱,esi源,正离子模式,分子式c28h33n5o4,理论值503.25,测试值503.58。元素分析(c28h33n5o4),理论值c:66.78,h:6.61,n:13.91,o:12.71,实测值c:66.80,h:6.60,n:13.92,o:12.68。元素分析测试仪品牌:美国加联,型号:ce-440。
化合物合成例4
结构式(1-4)所代表的化合物可根据下面的合成路线4来合成:
(1)化合物1-4-a的合成
于1l的三口圆底烧瓶中投入2,5-二氨基碘苯(23.40g,100毫摩尔)、et3n(50.60g,500毫摩尔)和200g甲苯,将体系升温到60℃,然后缓慢向体系内滴加4-硝基-3-氯苯甲酰氯(46.20g,210毫摩尔)和200g甲苯的混合液,约0.5小时滴加完成,然后将体系升温到80℃搅拌8至10小时,tlc跟踪,直至原料2,5-二氨基碘苯无剩余。然后将反应体系降至室温,用500ml*3的去离子水,水洗三次,上层有机相脱干得到淡黄色固体,加入500ml甲醇/水的混合液打浆,此悬浊液经过滤烘干以78%收率得到化合物1-4-a。
(2)化合物1-4-b的合成
于1l的三口圆底烧瓶中投入1-4-a(60.11g,100毫摩尔)、pd(pph3)4(2.31g,2毫摩尔)、cui(0.38g,2毫摩尔)、et3n(50.60g,500毫摩尔)和dmf溶剂200ml,在20-40℃搅拌30min,而后向体系内滴加炔丙基胺(6.06g,110毫摩尔)和100ml溶剂dmf的混合液,1-2小时滴加完毕,于20-40℃保温反应4-10小时。
反应结束后,向体系中加入500ml1,2-二氯乙烷和500ml浓度为1mol/l的盐酸溶液,有机相用500ml*3的去离子水,水洗三次,机相脱干得到淡黄色固体,收率61%。
(3)化合物1-4-c的合成
于1l的三口圆底烧瓶中投入1-4-b(52.83g,100毫摩尔)和300gdmf,降温至0-10℃,滴加溴乙酸叔丁酯(19.51g,100毫摩尔)的200g二氯乙烷溶液,滴加用时0.5-1h。滴加完毕后室温搅拌20-24h。反应结束后,加入1l乙酸乙酯,用500ml去离子水水洗,脱除乙酸乙酯,得到42.41g淡黄色固体,收率为66%。
(4)化合物1-4-d的合成
于1l的三口圆底烧瓶中投入1-4-c(64.25g,100毫摩尔)和300g二氯乙烷,降温至0-10℃,滴加入二碳酸二叔丁酯(21.83g,100毫摩尔)的200g二氯乙烷溶液,滴加用时0.5-1h。滴加完毕后室温搅拌24h。反应结束后,反应液用500ml去离子水水洗,脱除二氯甲烷,得到63.12g淡黄色固体,收率为85%。
(5)化合物1-4的合成
于1l的高压釜中投入获得的化合物1-4-d(37.13g,50毫摩尔)、5%的钯碳(4.13g,含水,固含量为45%)和600g四氢呋喃,将高压釜密封,用氢气置换3-5次后,氢气加压至0.5-1.0mpa,搅拌下于45-55℃反应。反应结束后,用孔径为0.2μm的滤膜将催化剂滤出,滤液脱溶剂,所得固体加入60g乙醇并搅拌30min,经过抽滤和烘干,将以90%的收率得到一种类白色的固体化合物1-4。
该化合物1-4的高分辩率质谱,esi源,正离子模式,分子式c34h41cl2n5o6,理论值685.24,测试值685.47。元素分析(c34h41cl2n5o6),理论值c:59.48,h:6.02,cl:10.33,n:10.20,o:13.98,实测值c:59.47,h:6.03,cl:10.35,n:10.21,o:13.99。元素分析测试仪品牌:美国加联,型号:ce-440。
化合物合成例5
结构式(1-5)所代表的化合物可根据下面的合成路线5来合成:
(1)化合物1-5-a的合成
于1l的三口圆底烧瓶中投入1,2-二氨基-3,5-二碘苯(35.99g,100毫摩尔)、et3n(50.60g,500毫摩尔)和200g甲苯,将体系升温到60℃,然后缓慢向体系内滴加4-硝基-3-异丙基苯甲酰氯(47.80g,210毫摩尔)和200g甲苯的混合液,约0.5小时滴加完成,然后将体系升温到80℃搅拌8至10小时,tlc跟踪,直至原料1,2-二氨基-3,5-二碘苯无剩余。然后将反应体系降至室温,用500ml*3的去离子水,水洗三次,上层有机相脱干得到淡黄色固体,加入500ml甲醇/水的混合液打浆,此悬浊液经过滤烘干以77%收率得到化合物1-5-a。
(2)化合物1-5-b的合成
于1l的三口圆底烧瓶中投入1-5-a(74.23g,100毫摩尔)、pd(pph3)4(2.31g,2毫摩尔)、cui(0.38g,2毫摩尔)、et3n(50.60g,500毫摩尔)和dmf溶剂200ml,在20-40℃搅拌30min,而后向体系内滴加炔丙基胺(6.06g,110毫摩尔)和100ml溶剂dmf的混合液,1-2小时滴加完毕,于20-40℃保温反应4-10小时。
反应结束后,向体系中加入500ml1,2-二氯乙烷和500ml浓度为1mol/l的盐酸溶液,有机相用500ml*3的去离子水,水洗三次,机相脱干得到淡黄色固体。
(3)化合物1-5-c的合成
于1l的三口圆底烧瓶中投入1-5-b(59.66g,100毫摩尔)和300g二氯乙烷,降温至0-10℃,滴加入二碳酸二叔丁酯(45.83g,210毫摩尔)的200g二氯乙烷溶液,滴加用时0.5-1小时。滴加完毕后室温搅拌24小时。反应结束后,反应液用500ml去离子水水洗,脱除二氯甲烷,得到68.53g淡黄色固体,收率为86%。
(5)化合物1-5的合成
于1l的高压釜中投入获得的化合物1-5-c(39.84g,50毫摩尔)、5%的钯碳(4.43g,含水,固含量为45%)和600g四氢呋喃,将高压釜密封,用氢气置换3-5次后,氢气加压至0.5-1.0mpa,搅拌下于45-55℃反应。反应结束后,用孔径为0.2μm的滤膜将催化剂滤出,滤液脱溶剂,所得固体加入80g乙醇并搅拌30min,经过抽滤和烘干,将以88%的收率得到一种类白色的固体化合物1-5。
该化合物1-5的高分辩率质谱,esi源,正离子模式,分子式c42h60n6o6,理论值744.46,测试值744.96。元素分析(c42h60n6o6),理论值c:67.72,h:8.12,n:11.28,o:12.89,实测值c:67.70,h:8.13,n:11.29,o:12.88。元素分析测试仪品牌:美国加联,型号:ce-440。
(二)聚合物的合成例
聚合物合成例1
在氮气氛围下,于500ml的三口圆底烧瓶中投入结构式(1-1)所代表的芳胺化合物(22.98g,50毫摩尔)(以下简称1-1),对苯二胺(2.16g,20毫摩尔)(以下简称2-1),4,4’-二氨基二苯乙烷(6.37g,30毫摩尔)(以下简称2-2)和144.84g的n-甲基-2-吡咯烷酮(以下简称nmp),将所得悬浮液搅拌直至得到一个黄色的溶液。然后将19.61g(100毫摩尔)的1,2,3,4-环丁烷四羧酸二酐(以下简称a-1)和144.84gnmp加入体系。反应放热,室温搅拌4小时,得到浓度为15%溶解在nmp中的聚酰胺酸聚合物。
聚合物合成例1至20及聚合物合成比较例1至10可通过与聚合物合成例1相似的方法制备,其浓度均为15%,溶剂均为nmp,其不同之处在于:所用单体的种类及用量有所改变,具体结果见下表1和表2,此处不再赘述。
在表1和表2中:
a-1:1,2,3,4-环丁烷四羧酸二酐
a-2:1,3-二甲基-1,2,3,4-环丁烷四羧酸二酐
a-3:2,3,5-三羧基环戊基乙酸二酐
1-1:由式(1-1)表示的化合物
1-2:由式(1-2)表示的化合物
1-3:由式(1-3)表示的化合物
1-4:由式(1-4)表示的化合物
1-5:由式(1-5)表示的化合物
2-1:对苯二胺
2-2:4,4’-二氨基二苯乙烷
2-3:4,4′-二氨基二苯醚
2-4:2,2’-二甲基-4,4’-二氨基联苯
2-5:1,3-双(4-氨基苯氧基)丙烷
2-6:由式(2-6)表示的化合物
表1合成例各聚合物所用单体种类及用量
表2比较合成例各聚合物所用单体种类及用量
(三)液晶取向剂、液晶取向膜及液晶显示元件的实施例与比较例
实施例1
a、液晶取向剂
氮气保护下,于三口圆底烧瓶中投入100重量份的聚合物(聚合物合成例1)、86重量份的nmp和114重量份的乙二醇单丁醚,体系于室温下搅拌60分钟,然后用0.2μm的滤膜过滤该溶液形成实施例1的液晶取向剂,其浓度为5%,溶剂为nmp:bc=60:40。
b、液晶取向膜及液晶显示元件
本发明实施例中仅以ips和ffs型液晶显示元件来解释本发明,但本发明不限于此。
实施例1的液晶取向剂用旋涂的方式涂覆在一片具有ips(面内切换型)或ffs(边缘场切换型)ito电极的第一玻璃基板上,以形成预涂层。经过预固化(热板,85℃,10分钟),主固化(循环烘箱,225℃,50分钟),曝光(254nm偏振光、5mw/cm2、500mj/cm2)将得到具有ito电极上面形成实施例1的液晶取向膜的第一玻璃基板。
用旋涂的方式将实施例1的液晶取向剂涂覆在一片不具有ito电极的第二玻璃基板上,以形成预涂层。也经过上述预固化、主固化、爆光后得到上面形成实施例1的液晶取向膜的第二玻璃基板。
将一种密封胶涂布在第一玻璃基板的周边,并留有液晶液入口,将3.5μm的间隔子洒在另一片基板上。然后将这两片玻璃基板以与取向方向为反平行的方式进行贴合(5kg,30min),然后将密封胶固化,接着将液晶注入,然后利用紫外光硬化胶封住液晶注入口,并以紫外光使紫外光硬化胶硬化,再分别于两片玻璃基板的外侧贴上偏光板,即可获得实施例1的ips或ffs型液晶显示元件。
将未形成液晶取向膜的透明玻璃基板的透过率设为100%,对形成有液晶取向膜的基板进行uv-vis光谱,测定结果示于图1中,紫外可见分光光度计型号为cary100(安捷伦公司制造)。根据测试结果计算出390nm-550nm的透过率的平均值作为液晶取向膜的透过率,结果为95.8%。
对实施例1的液晶显示元件进行评价,结果见表3。
实施例2至20
液晶取向剂、液晶取向膜及液晶显示元件的实施例2至实施例20可通过与实施例1相同的步骤制备,不同之处在于:所用聚合物替换为实施例2-20中所对应的聚合物,所制备的液晶取向剂的浓度均为5%,溶剂比例均为nmp:bc=60:40,对实施例2-20的液晶显示元件进行评价及结果见表3。
比较例1至12
液晶取向剂、液晶取向膜及液晶显示元件的比较例1至比较例12可通过与实施例1相同的步骤制备,不同之处在于所用聚合物替换为比较例1-12中所对应的聚合物,其对应液晶取向剂的浓度均为5%,溶剂比例均为nmp:bc=60:40。除变更所使用的聚合物以外,依据实施例1来进行uv-vis光谱的测定。将比较例1的uv-vis光谱示于图2中,结果为85.9%。比较例1-12所对应的液晶显示元件的评价结果见表3。
表3
评价方法
(1)透过率的表征
将未形成液晶取向膜的透明玻璃基板的透过率设为100%,对形成有液晶取向膜的基板进行全波长(200-780nm)透过率测试,紫外可见分光光度计型号为cary100(安捷伦公司制造)。根据测试结果计算出390nm-550nm的透过率的平均值,将透过率为80-90%判断为不良,将90-95%判断为一般,将95%以上判断为优秀。
(2)对比度的表征
将所述制造的液晶显示元件(未贴合偏光板的液晶单元)以饱和交流电压驱动30小时后测试对比度。仪器为亮度计(装置型号:3298f,制造商:yokogawa),在配备正交尼科尔棱镜(crossnicol)的偏光显微镜下测试液晶显示元件,测试的最小亮度作为黑度。对液晶原件施加任意矩形波电压,测试最大亮度作为白度。对比度定义为白度与黑度的比值。
液晶显示元件对比度评价结果如下:
√:对比度≥1800,液晶显示元件对比度优秀
○:1200≤对比度<1800,液晶显示元件对比度一般
x:对比度<1200,液晶显示元件对比度差
由此可见,与现有技术相比,本发明的液晶取向剂由于所用芳胺单体中含有特定结构的芳胺化合物b-1,因此本发明的液晶取向剂所制得的液晶取向膜具有成膜透过率高和所制得的液晶显示元件具有对比度高的优点,并且本发明的实施方法简单,市场前景广阔,适合规模化应用推广。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



