
 商标分类
商标分类  商标转让
商标转让 
4-羟基吡啶酮类化合物及其制备方法和用途与流程
 2021-02-02 22:02:24|
2021-02-02 22:02:24| 590|
590| 起点商标网
起点商标网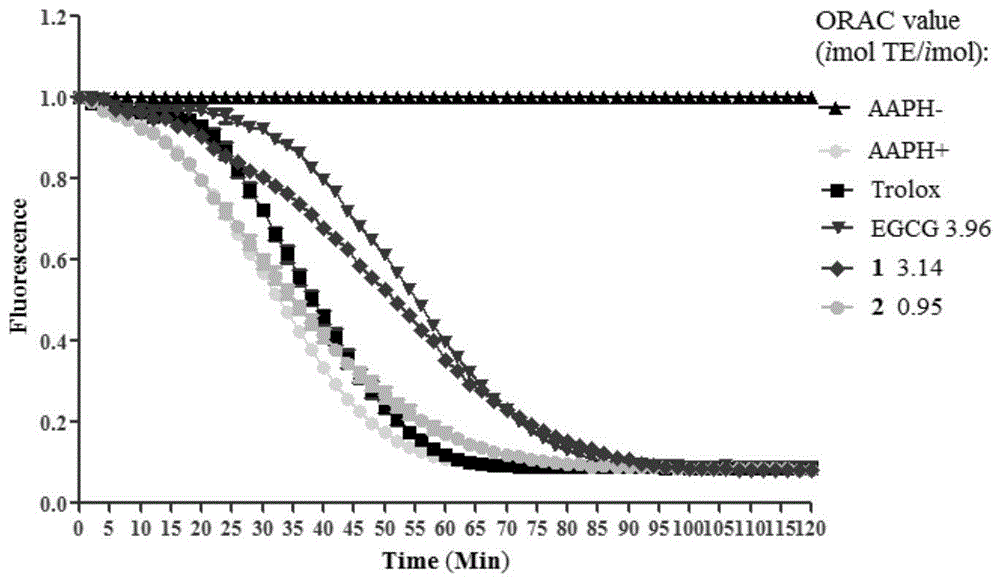
本发明属于天然产物技术领域,具体涉及4-羟基吡啶酮类化合物及其制备方法和用途。
背景技术:
生物体内的氧化指失去电子或得到氧原子的过程,会升高生物分子、分子中的原子或离子的氧化态,并在生物体内形成自由基。人体氧化还原过程的失衡会导致生成过量的自由基,进而引发机体的一系列抗氧化防御反应,这个过程称为氧化应激。氧化应激与诸多疾病的发生发展相关,如冠心病、动脉粥样硬化、帕金森氏病、脑卒中等等。因此,抗氧化剂和自由基清除试剂在医药领域有着广泛的应用。
真菌来源的天然小分子化合物是药物先导化合物的重要来源。真菌在长期适应环境或经历共生、寄生的过程中,进化出了独特的次级代谢途径,因而真菌次级代谢产物具有特异的化学结构和多样的生物活性,真菌来源的天然产物已有很多成药的先例。因此,从真菌次级代谢产物中寻找新的天然活性化合物是重要的药物发现方式。
弯颈霉属(tolypocladiumsp.)是w.gams于1971年建立的新属,该属的特点是瓶梗基部呈球形或椭圆形的膨大,瓶颈细长而弯曲,分生孢子单生或黏聚成小头状,至目前国际真菌名录数据库中记录了弯颈霉属有43种。弯颈霉属是一类重要虫生真菌,生态分布广,寄主种类多,弯颈霉属很多种被发现是虫草属无性型,与虫草属的关系十分密切,2014年国际上的一些著名真菌学家基于分子生物学的新分类系统将弯颈霉属(tolypocladiumsp.)归入到线虫草科(ophiocordicipitaceae),更为重要是弯颈霉属能产生多种杀虫和抗菌的有效成分,如环孢菌素(cyclosporin)、大团囊素(ophiocordin,蛋白酶强抑制剂)、线肽素(efrapeptin)、ophiosetin(四聚胺酸素)、多种杀虫剂等。
技术实现要素:
发明目的
本发明的目的在于对弯颈霉属(tolypocladiumsp.)真菌的发酵提取物进行系统制备和分离并鉴定,寻找具有抗氧化和清除自由基功能的药物或先导化合物。
技术方案
本发明的第一方面,提供了一种4-羟基吡啶酮类化合物或其药学上可接受的盐、其溶剂化物或其代谢产物,其特征在于,所述4-羟基吡啶酮类化合物如式(i)所示:
其中,r为h、任选取代的c1-c4烷基、任选取代的c1-c4烷氧基、-oh、对羟基苯基(即
进一步的,所述化合物具有以下如式1或式2所示结构:
进一步的,所述药学上可接受的盐为所述4-羟基吡啶酮类化合物与化学上可接受的酸或碱反应制得的盐;更进一步的,所述化学上可接受的酸为无机酸或有机酸;进一步优选的,所述无机酸选自盐酸、硫酸、硝酸、氢溴酸中的任意一种;或进一步优选的,所述有机酸选自乙酸、丙酸、丙二酸、丁酸、乳酸、甲磺酸、乙磺酸、苯磺酸、对甲苯磺酸、马来酸,苯甲酸、琥珀酸、苦味酸、酒石酸、柠檬酸、富马酸中的任意一种;
或更进一步的,所述药学上可接受的盐为所述4-羟基吡啶酮类化合物与化学上可接受的碱进行反应制得的盐;更进一步的,所述化学上可接受的碱为无机碱或有机碱;进一步优选的,所述无机碱选自氢氧化钠、氢氧化钾、氢氧化钙、碳酸钠、碳酸钾、碳酸氢钠、碳酸氢钾中的任意一种;或进一步优选的,所述有机碱选自三甲胺、三乙胺、吡啶中的任意一种;
进一步优选的,所述药学可接受的盐可以为钾盐、钠盐、铵盐、钙盐、吡啶盐、胆碱盐中任意一种或几种;
本发明的第二方面提供了一种上述任一种4-羟基吡啶酮类化合物或其药学上可接受的盐、其溶剂化物或其代谢产物的制备方法,包括:由弯颈霉属(tolypocladiumsp.)真菌经微生物发酵后获得提取物,再采用色谱法将所述提取物分离后得到;
进一步的,所述4-羟基吡啶酮类化合物或其药学上可接受的盐、其溶剂化物或其代谢产物的制备方法,包括以下步骤:
s1:将弯颈霉属(tolypocladiumsp.)真菌菌种接种于培养基中进行培养,得到发酵物;
s2:将步骤s1所得发酵物用70~95%v/v乙醇浸泡10~15h,然后超声提取1~3次,每次10~30min,合并提取液浓缩至干燥,得粗提物;
s3:将步骤s2所得粗提物经硅胶柱色谱、制备高效液相色谱(即制备hplc)分离并鉴定,即得;
进一步的,s1所述的操作包括以下步骤:将所述菌株经马铃薯葡萄糖琼脂斜面培养基(即pda斜面)活化后,10~30℃下,在马铃薯葡萄糖液体培养基(即pdb培养基)中培养3~7天,然后接种到大米培养基中,10~30℃下,静置培养24~60天得到发酵物;优选的,在pda和pdb中的培养温度均为25℃;优选的,在pdb中培养7天;优选的,在大米培养基中静置培养60天;
进一步的,s2中所述乙醇浓度为95%v/v;
进一步的,s2中所述浸泡时间为12h;
进一步的,s2中所述超声提取次数为3次、提取时间为30min;
进一步的,s3中所述经硅胶柱色谱分离的操作具体包括:将步骤s2所得粗提物经硅胶柱分离,采用二氯甲烷-甲醇溶液梯度洗脱,依次用甲醇:二氯甲烷体积比为1:99,2:98,5:95,10:90,15:85,20:80,25:75,30:70,50:50,100:0进行洗脱,得到fr.1-12共12个馏分,其中第9个子馏分即以甲醇:二氯甲烷体积比为50:50进行洗脱获得的馏分fr.9;
进一步的,s3中所述经制备hplc分离并鉴定的操作具体包括:以甲醇:水的体积比为75:25配制成溶液作为流动相对馏分fr.9进行等度洗脱,所述流动相流速为3.0ml/min,以紫外检测器进行检测,所述uv检测波长为210nm或280nm,得到化合物1为tolypyridonec,其tr为32.0min;以及化合物2为tolypyridoned,其tr为26.9min;
更进一步的,所述制备hplc采用的色谱柱为cosmosil5c18,规格为φ10.0×250mm,填料粒径5μm);
更进一步的,所述制备hplc采用的柱温为30℃;
本发明的第三方面提供了一种药物组合物,包括如上任一项所述的4-羟基吡啶酮类化合物或其药学上可接受的盐、其溶剂化物或其代谢产物;
进一步的,所述药物组合物中所述4-羟基吡啶酮类化合物或其药学上可接受的盐、其溶剂化物或其代谢产物占所述药物组合物质量的0.01~90%;优选的,所述药物组合物中所述4-羟基吡啶酮类化合物或其药学上可接受的盐、其溶剂化物或其代谢产物占所述药物组合物质量的0.1~20%;进一步优选的,所述药物组合物中4-羟基吡啶酮类化合物或其药学上可接受的盐、其溶剂化物或其代谢产物占所述药物组合物质量的1~10%;
进一步的,尽管本发明的化合物可以不经任何配制直接给药,但优选与药学上可接受的载体制备成药物制剂使用;进一步优选的,所述药学上可接受的载体包括稀释剂、润滑剂、粘合剂、崩解剂、稳定剂、溶剂;更进一步的,本发明所述稀释剂包括但不限于淀粉、微晶纤维素、蔗糖、糊精、乳糖、糖粉、葡萄糖等;更进一步的,所述润滑剂包括但不限于硬脂酸镁、硬脂酸、氯化钠、油酸钠、月桂醇硫酸钠、泊洛沙姆等;更进一步的,所述粘合剂包括但不限于水、乙醇、淀粉浆、糖浆、羟丙基甲基纤维素、羧甲基纤维素钠、海藻酸钠、聚乙烯吡咯烷酮等;更进一步的,所述崩解剂包括但不限于淀粉、碳酸氢钠和/或枸橼酸、酒石酸、低取代羟丙基纤维素等;更进一步的,所述稳定剂包括但不限于多糖如金合欢胶、琼脂、藻酸、纤维素醚、羧甲基甲壳酯等;更进一步的,所述溶剂包括但不限于水、平衡的盐溶液等;
进一步的,所述药物组合物为口服制剂或注射剂;进一步优选的,所述口服制剂选自普通片剂、分散片、肠溶片、颗粒、胶囊、滴丸、散剂、口服液、乳剂中的任意一种;或进一步优选的,所述注射剂为小水针剂、输液剂或冻干粉针;
本发明的第四方面提供了一种如上述任一项所述的4-羟基吡啶酮类化合物或其药学上可接受的盐、其溶剂化物或其代谢产物或药物组合物在制备氧化应激相关疾病药物中的应用;
进一步地,所述氧化应激相关疾病包括但不限于冠心病、动脉粥样硬化、帕金森氏病或脑卒中。
附图说明
图1化合物1和2的氧自由基吸收能力图
有益效果
本发明通过对弯颈霉属(tolypocladiumsp.)真菌提取物的分离和结构鉴定,获得了两个新化合物,并对这两个新化合物进行药理活性测试,发现其具有良好的自由基清除活性,具有抗氧化作用,能够用于制备治疗氧化应激相关疾病的药物,具有良好的药学应用前景。
具体实施方式
下面将详细举例说明本发明。需要指出的是,所述实施例说明了一些制备或使用方法,然而,要理解的是,这些实施例不限制本发明。本发明的保护范围以所附权利要求书记载的内容为准。
1.菌株来源
来自于弯颈霉属(tolypocladiumsp.)真菌。
2.实验仪器
eyela旋转蒸发仪n-1001(东京rikakikai公司);dionex分析型和制备型高效液相色谱仪(美国thermofisherscientific公司);cosmosil5c18分析色谱柱(10×250mm,5μm)和制备色谱柱(20×250mm,5μm)(日本nacalaitesque公司);jascov-550紫外测定仪购自日本jascointernational公司;jascoft/ir-480plus红外测定仪购自日本jascointernational公司;jascop-1020旋光测定仪购自日本jascointernational公司;brukerav-600mhz核磁共振仪购自瑞士brukerbiospin公司;waterssynaptg2tof高分辨质谱仪购自美国waters公司。
3.实验材料
分析纯环己烷、乙酸乙酯、乙醇购自天津市富宇精细化工有限公司;氘代二甲基亚砜购自德国sigma-aldrich公司;色谱级甲醇购自山东禹王实业有限公司;ods(50μm)购自日本ymc公司;薄层色谱用硅胶gf254和柱色谱硅胶(200-300目)均为青岛海洋化工厂产品;
以下缩写贯穿整个说明书:
tr:保留时间
dmso-d6:氘代二甲基亚砜
δh:氢的化学位移
δc:碳的化合物位移
实施例1:弯颈霉属(tolypocladiumsp.)真菌的发酵和提取
(1)tolypocladiumsp.菌种经pda斜面活化后接种于pdb培养基,在25℃下以200r·min-1震荡培养3d制备种子液,再按照5ml/瓶接种量把种子液接种到20瓶装有大米培养基(所述大米培养基由以下组分组成:大米70g/瓶,纯净水110ml/瓶)的三角烧瓶中,25℃下静置培养60天,得到发酵物;
(2)将发酵物用95%乙醇浸泡12h,超声提取3次(每次30分钟),提取液减压浓缩至干,得到粗提物(20.1g)
实施例2:化合物1(tolypyridonec)和2(tolypyridoned)的制备
(1)上述粗提物使用硅胶柱色谱进行分离,使用二氯甲烷-甲醇溶液梯度洗脱,依次用甲醇:二氯甲烷体积比1:99,2:98,5:95,10:90,15:85,20:80,25:75,30:70,50:50,100:0进行洗脱,得到fr.1-12共12个馏分。
(2)将子馏分fr.9经hplc制备,色谱柱为cosmosil5c18,规格为φ10.0×250mm,填料粒径5μm),柱温为30℃;以甲醇-水溶液为流动相进行洗脱,甲醇:水的体积比为75:25,流速为3ml/min,以紫外检测器进行检测,所述uv检测波长为210nm或280nm,得到化合物1为tolypyridonec(tr为32.0分钟,约30毫克),化合物2为tolypyridoned(tr为26.9分钟,约27毫克)。
实施例3:
本实施例中化合物1和2的结构鉴定结果如下:
化合物1
淡黄色针状结晶,熔程168-171℃;
化合物2
淡黄色油状物;
表1化合物1和2的核磁共振图谱(dmso-d6,600mhzfor1hand150mhzfor13c,jinhz)
实施例4:化合物1和2的氧自由基吸收能力测试
(1)实验原理
氧自由基吸收能力测试以α-生育酚的水溶性类似物-trolox(6-羟基-2,5,7,8-四甲基苯并二氢吡喃-2-羧酸)为标准对照,测定物质总抗氧化能力。荧光物质disodiumfluorescein在485nm光的激发下,可发射527nm的荧光。aaph[2,2'-偶氮(二眯基丙烷)二氯化氢]能释放过氧自由基(roo·),并将disodiumfluorescein(fl)氧化,从而荧光特性消失。抗氧化剂可与disodiumfluorescein竞争氧化剂,使其荧光消退的速度减缓。根据这种特性测定化合物的氧自由基吸收能力。
75mmph7.4磷酸盐缓冲液(pbs):分别称取5.103gkh2po4,8.56gk2hpo4·3h2o,混合,加去离子水至500ml,用1mol/lhcl调节ph至7.4。
disodiumfluorescein(fl)溶液:取0.237mgfl,用1ml上述磷酸钾缓冲液配成0.63mm的母液,置于4℃避光保存。临用时用上述磷酸钾缓冲液稀释1000倍,配成630nm的工作液。
trolox溶液:取1mgtrolox,加入上述磷酸钾缓冲液199.8μl,得到0.02m的母液。取母液1μl,加入缓冲液799μl稀释为25μm的溶液,再取25μm的溶液50μl,加入缓冲液50μl,得到100μl12.5μm的工作液。
aaph溶液:取0.248gaaph,溶解于50ml上述磷酸钾缓冲液,得到18.3mm的工作液。
受试单体化合物1和2:取适量单体化合物,分别配制成2×10-2mol/l二甲基亚砜溶液作为母液。取母液0.5μl,加入缓冲液799.5μl稀释为12.5μm的工作液。
(2)实验方法
实验共分为5个组:空白对照组,阴性对照组,标准对照组,阳性药组和受试药物组,所有测试皆为4个重复,详细实验设计如下表2所示。
表2详细实验设计
(3)实验结果
实验结果的计算和比较参考amorati等人发表的方法[1]。如图1所示,以527nm处荧光值为横坐标,以时间为纵坐标绘制氧自由基吸收能力图,化合物1的氧自由基吸收能力(3.14)与阳性药表没食子儿茶素没食子酸酯(epigallocatechingallate,egcg3.96)相当,化合物2(0.95)也表现出了相应的活性,只是程度不如化合物1。
在本说明书的描述中,参考术语“实施例”、“一些实施方案”或“示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明构思的前提下,还可以做出若干改进和修饰,这些改进和修饰也应视为本发明的保护范围内。
参考文献
[1]amorati,r.;valgimigli,l.,advantagesandlimitationsofcommontestingmethodsforantioxidants.freeradicalresearch2015,49,(5),633-649.
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



