
 商标分类
商标分类  商标转让
商标转让 
抗病毒药物Musellarin及类似物分子、制备方法和应用与流程
 2021-02-02 21:02:23|
2021-02-02 21:02:23| 350|
350| 起点商标网
起点商标网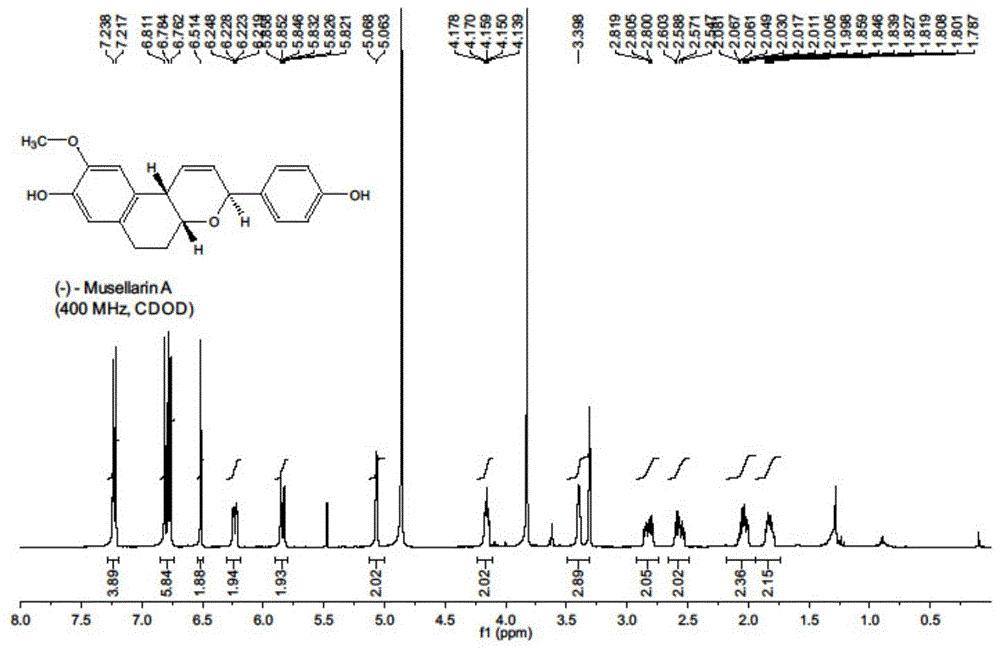
本发明属于药物化学技术领域,具体涉及一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子、制备方法和应用。
背景技术:
禽流感是一类由禽流感病毒感染引起的传染性疾病,禽流感病毒具有多种亚型,且极易发生变异。目前常见的禽流感病毒亚型主要包括h5n1、h1n1、h5n2、h7n3、h7n7、h9n2和h7n9亚型。其中,h7n9病毒是最新发现的一种流感亚型。其高达40%的致死率引起了全球范围的高度关注,研究学者开始积极研究关于该类疾病的药物。
目前已报导的抗流感药物根据其作用靶点可分为四大类。第一类为金刚烷胺和乙胺,此类药物可抑制病毒的m2蛋白,导致病毒不能脱壳入胞,从而阻止流感病毒的释放。然而该类靶点药物仅对甲型流感有效,并且发现流感病毒对此类药物产生了耐性。第二类为神经氨酸酶抑制剂,作用靶点是病毒包膜上的神经氨酸酶,该药物可以抑制病毒的神经氨酸酶,阻止子代病毒从感染细胞内释放,从而起到杀死病毒的效果。目前广为使用的药物为奥司他韦,扎那米韦和帕拉米韦。其中,扎那米韦不利于生物吸收,利用率低;帕拉米韦是第一个可静脉注射的神经氨酸酶抑制剂;奥司他韦在扎那米韦的基础上开发而成,奥司他韦能够有效治疗甲型流感和乙型流感,该药已获得fda的批准,2002年获准进入中国,作为治疗重度流感的有效药物。此外,奥司他韦具有副作用,包括行为异常,出现幻觉和视觉障碍等,其安全性受到学者的质疑。第三类药物为法匹拉韦,也称为t-705,其结构类似嘧啶。t-705进入细胞后,会被细胞的酶进行三磷酸化,变成其活性成分,因其结构类似三磷酸核苷gtp,进而可影响病毒rna的复制与转录过程。第四类药物为2018年日本上市的新型抗流感药物xofluza。不同于以往的药物,该药作用的可能靶点为rna聚合酶中的pa内切酶。pa内切酶可从宿主细胞前体的mrna中通过“cap-snatching”的方式剪切获得mrna的冒状结构,用于流感病毒自身的合成。“cap-snatching”是流感病毒复制周期中的关键环节,阻断这一环节,将可以选择性的阻断流感病毒的转录过程。此药物的获批也证明可将流感病毒的rna聚合酶作为新的药物靶点。
目前获卫生部推荐的药物为神经氨酸酶抑制剂,但是其副作用包括幻觉和行异常引起了人们重视。2017年月报道了3株h1n1对神经氨酸酶抑制剂敏感性高度降低,耐药出现使人类再一次暴露在流感病毒的威胁当中。针对多种亚型和耐药性,需要更为有效的靶点以及针对新靶点设计的新型药物。因此,开发有效的抗流感病毒药物,显得尤为紧迫。
技术实现要素:
本发明的目的在于克服现有技术的上述不足,提供一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子及其制备方法和应用,旨在解决现有流感病毒药物种类有限,副作用大的技术问题。
为实现上述发明目的,本发明采用的技术方案如下:
本发明一方面提供一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子,其化学结构通式如式i所示:
其中,r1选自羟基或烷氧基,r2选自羟基或烷氧基,r3自氢原子、羟基或烷氧基,r4选自羟基或烷氧基。
本发明提供的以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子,其分子结构可以流感病毒rna聚合酶为靶点,通过抑制流感病毒的rna聚合酶的活性,直接阻断流感病毒的转录和复制过程,本发明通过实验证明,该式i所示的分子可有效抑制rna聚合酶的活性,对流感病毒的rna聚合酶为靶点具有高效抑制性和高选择性,相比商品药物法匹拉韦t-705,具有更强的抑制效果和较低的细胞毒性,因此,具有开发为抗流感病毒药物分子的潜力。
本发明另一方面提供一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子的制备方法,包括如下步骤:
将式4所示的糠醇底物进行achmatowicz重排反应,然后进行乙酰化反应,得到式5所示的化合物;
将式5所示的化合物进行脱氧反应,得到式6所示的化合物;
将式6所示的化合物分别和式7-9所示的重氮盐进行偶联反应,分别得到式10-12所示的化合物;
将式10-12所示的化合物依次进行还原反应、分子内傅克闭环反应和脱保护反应;
本发明提供制备方法,工艺简单成本低,其制备得到的抗病毒药物musellarin及类似物分子可以流感病毒rna聚合酶为靶点,通过抑制流感病毒的rna聚合酶的活性,直接阻断流感病毒的转录和复制过程,因此具有开发为抗流感病毒药物分子的潜力。
最后,本发明提供一种如式i所示的抗病毒药物musellarin及类似物分子和/或本发明所述的制备方法得到的抗病毒药物musellarin及类似物分子在制备抗病毒药物中的应用。
附图说明
图1是本发明实施例提供的式1所示化合物的1h-nmr图谱;
图2是本发明实施例提供的式1所示化合物的13c-nmr图谱;
图3是本发明实施例提供的式2所示化合物的1h-nmr图谱;
图4是本发明实施例提供的式2所示化合物的13c-nmr图谱;
图5是本发明实施例提供的式3所示化合物的1h-nmr图谱;
图6是本发明实施例提供的式3所示化合物的13c-nmr图谱;
图7是本发明实施例提供的式1、式2、式3与流感病毒rna聚合酶结合位置示意图;
图8是本发明实施例提供的式1、式2、式3细胞毒性lc50的示意图;
图9是本发明实施例提供的式1、式2、式3抑制效果ic50的示意图;
图10是本发明实施例提供的式1抑制流感病毒聚合酶体外转录的示意图。
具体实施方式
为了使本发明要解决的技术问题、技术方案及有益效果更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
一方面,本发明实施例提供了一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子,化学结构通式如式i所示:
其中,r1选自羟基或烷氧基,r2选自羟基或烷氧基,r3自氢原子、羟基或烷氧基,r4选自羟基或烷氧基。
本发明实施例提供的一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子,其分子结构可以流感病毒rna聚合酶为靶点,通过抑制流感病毒的rna聚合酶的活性,直接阻断流感病毒的转录和复制过程,本发明通过实验证明,该式i所示的分子可有效抑制rna聚合酶的活性,对流感病毒的rna聚合酶为靶点具有高效抑制性和高选择性,相比商品药物法匹拉韦t-705,具有更强的抑制效果和较低的细胞毒性,因此,具有开发为抗流感病毒药物分子的潜力。
流感病毒的rna聚合酶负责rna的转录与复制,是流感病毒活性的核心成份,有效抑制rna聚合酶的活性可阻断流感病毒的复制。不同于人体内的dna转录与复制过程,针对rna聚合酶的药物不会对人体产生伤害。由于甲型、乙型流感病毒的rna聚合酶高度的遗传序列保守性,特别在关键活性位置的序列相似性,病毒的rna聚合酶被称为药物的“超级靶标”。不同于以往的神经氨酸酶为靶点,病毒rna聚合酶作为药物靶点时,由于聚合酶具有很强的遗传保守性,将使病毒不易对药物产生抗药性,且该类药物将对具有此种rna聚合酶的一系列禽流感病毒表现出广谱抑制性;同时,因为病毒rna聚合酶的活性位点特异性,与其作用的药物将不会影响人体细胞的正常功能,具有更低的细胞毒性。因此,基于上述,本发明实施例提供了一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子。
具体地,r1中的烷氧基为甲氧基、乙氧基、丙氧基、丁氧基和戊氧基中的至少一种;r2中的烷氧基为甲氧基、乙氧基、丙氧基、丁氧基和戊氧基中的至少一种;r3中的烷氧基为甲氧基、乙氧基、丙氧基、丁氧基和戊氧基中的至少一种;r4中的烷氧基为甲氧基、乙氧基、丙氧基、丁氧基和戊氧基中的至少一种。
在一个实施例中,r1为甲氧基,r2为羟基,r3为氢原子,r4为羟基,如下式1所示的结构;或者,r1为甲氧基,r2为羟基,r3为甲氧基,r4为羟基,如下式2所示的结构;或者,r1为甲氧基,r2为羟基,r3为羟基,r4为甲氧基,如下式3所示的结构。
另一方面,本发明实施例还提供了一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子的制备方法,包括如下步骤:
s01:将式4所示的糠醇底物进行achmatowicz重排反应,然后进行乙酰化反应,得到式5所示的化合物;
s02:将式5所示的化合物进行脱氧反应,得到式6所示的化合物;
s03:将式6所示的化合物分别和式7-9所示的重氮盐进行偶联反应,分别得到式10-12所示的化合物;
s04:将式10-12所示的化合物依次进行还原反应、分子内傅克闭环反应和脱保护反应;
本发明实施例提供的一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子的制备方法,工艺简单成本低,其制备得到的分子可以流感病毒rna聚合酶为靶点,通过抑制流感病毒的rna聚合酶的活性,直接阻断流感病毒的转录和复制过程,因此具有开发为抗流感病毒药物分子的潜力。
本发明实施例提供的式1所示有机小分子、式2所示有机小分子、式3所示有机小分子,可以通过上述制备方法合成,也可以从植物中提取获得。进一步优选地,所述式1-3所示化合物的制备方法包括以下步骤:
s01.提供式4所示糠醇底物,在过硫酸氢钾制剂、溴化钾和碳酸氢钠下进行achmatowicz重排反应,制备得到吡喃酮中间体,然后在乙酸酐和碱的作用下发生乙酰化反应,制备得到式5所示化合物;
s02.将式5所示化合物在锌和乙酸下发生脱氧反应,制备得到式6所示化合物;
s03.将式6所示化合物和重氮盐(式7-9)经钯催化偶联反应,分别制备得到式10-12所示化合物;
s04.将式10-12所示化合物(注:式10所示化合物需先乙酰化把羟基保护)先经过硼氢化钠还原,并在三氟甲磺酸酐的作用下进行分子内傅克闭环反应,接着进行脱保护反应,制备得到式1-3所示药物分子。
本发明实施例提供的一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子的制备方法,以式4所示糠醇化合物为原料,在过硫酸氢钾制剂、溴化钾和碳酸氢钠下进行重排反应,然后进一步将所得的吡喃酮中间体,经乙酸酐的作用下发生乙酰化反应,制备得出式5所示化合物。该方法简单易控,且得到高产物收率和高ee值。
具体优选的,上述步骤s01中,以式4所示糠醇化合物为原料,在过硫酸氢钾制剂、溴化钾和碳酸氢钠下进行重排反应,然后进一步将所得的吡喃酮中间体,经乙酸酐的作用下发生乙酰化反应,制备得出式5所示化合物的反应式如下所示:
上述步骤s02中,将式5所示二氢吡喃在还原剂和乙酸的作用下发生γ脱氧反应,制备得到式6所示分子。优选的,所述还原剂为锌粉,由此可以得到高产量(86%)的式6所示分子。优选的,在这个温和的反应条件下,产物不容易差向异构化,产物能够保持高的ee值。相对一般的脱氧反应如使用二碘化钐作还原剂,本发明实施例这条件的重复性较高,特别是在放大化学反应规模下,也能得到高产率和高ee值。
具体优选的,上述步骤s02中,以式5所示二氢吡喃在锌粉和乙酸的作用下发生γ脱氧反应,制备得到式6所示分子的反应式如下所示:
上述步骤s03中,将式6所示化合物经钯催化偶联反应分别制备得到式10-12所示化合物,优选的,所述钯催化剂为pd(oac)2,反应溶剂为乙腈,在乙酸钠的作用下与重氮盐(式7-9)反应,由此可以制备得到高产率(77-93%)的式10-12所示单一非对映异构体的2,6–trans--吡喃酮。在改反应条件中,不会引起任何因偶联产生的偶构化,而且它有利于β-氢化消除反应得出热力学上有利的共轭烯酮,从而制备得出式10-12所示的吡喃酮。
具体优选的,上述步骤s03中,以式6和重氮盐(式7-9)所示二氢吡喃经钯催化偶联反应分别制备得到式10-12所示化合物的反应式如下所示:
上述步骤s04中,将式10-12所示化合物(式10所示化合物需先乙酰化把羟基保护)先经过还原剂把烯酮还原,并在三氟甲磺酸酐的作用下进行分子内傅克闭环反应,接着进行脱保护反应,制备得到式1-3所示药物分子。优选的,所述还原剂为硼氢化钠,由此得出1:1的异构体烯醇,这个步骤中的非对映体比例并不影响分子内傅克闭环反应后产物的dr值。优选的,闭环反应的温度为-78℃。该反应中,优选采用三氟甲磺酸酐,有利生成碳正离子中间体从而促进闭环反应的发生。然后,将闭环产物在四正丁基氟化铵和碳酸钾/甲醇的作用下,进行脱保护反应,制备得到式1-3所示的化合物。
将式10-12所示化合物先经过硼氢化钠还原得到,并在三氟甲磺酸酐的作用下进行分子内傅克闭环反应,接着进行脱保护反应,制备得到式1-3所示药物分子的反应式如下所示:
最后,本发明实施例提供一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子和/或上述制备方法得到的抗病毒药物musellarin及类似物分子在制备抗病毒药物中的应用。
具体地,所述抗病毒药物为以rna聚合物为靶点的抗流感病毒药物。
该抗病毒药物musellarin及类似物分子以甲型流感病毒rna聚合酶为靶点,可作为治疗甲型流感的抑制剂或者有潜力的药物前体分子使用,通过细胞实验证明了其毒性较低,而抑制效果强于商品药物法匹拉韦有效成分的t-705,因此可以开发为一类口服治疗流感的抗病毒药物。所述抗病毒药物包含上述式i所示结构的化合物,或所述抗病毒药物包含上述式i所示结构一种作为药物前体的化合物。
优选地,本发明实施例中,以甲型流感病毒rna聚合酶为靶点的抗病毒药物中,可以含有式1所示的有机小分子或者以式1所示的有机小分子作为药物前体化合物,也可以含有式2所示的有机小分子或者以式2所示的有机小分子作为药物前体化合物,也可以含有式3所示的有机小分子或者以式3所示的有机小分子作为药物前体化合物当然,也可以同时含有式1、式2、式3,或者同时以式1、式2、式3小分子作为药物前体化合物。
本发明先后进行过多次试验,现举一部分试验结果作为参考对发明进行进一步详细描述,下面结合具体实施例进行详细说明。
实施例1式1-式3所示化合物的制备
(1)一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子,分子结构如式1所示,其制备方法包括以下步骤:
提供式4所示糠醇化合物4.01g,四氢呋喃20ml/水5ml作反应介质,在0℃下添加过硫酸氢钾制剂3.65g、溴化钾118mg和碳酸氢钠416mg,在0℃下反应1-2小时,用碳酸氢钠淬灭反应,萃取浓缩得到粗产物,然后把粗产物加到dcm20ml中,在0℃下添加乙酸酐1.52g和三乙氨2.02,并在0℃下反应4小时,用氯化铵水溶液淬灭反应,萃取浓缩得到粗产物,通过柱层析分离得到式5所示二氢吡喃4.1g。
取式5所示的二氢吡喃1.2g置于烧瓶中,加入乙酸10ml和锌粉0.84g,在室温下反应1.5小时,反应结束后使用碳酸钾水溶液淬灭,使用dcm萃取并浓缩得到粗产物,通过柱层析分离得到式6所示分子0.9g。
取式6所示分子114mg置于烧瓶中,加入1.4mlacn,添加式7所示重氮盐70mg和乙酸钠69mg,氮气饱和15分钟,然后加入pd(oac)29.4mg,在室温下反应4小时,反应结束后使用氯化铵水溶液淬灭反应,萃取浓缩得到粗产物,通过柱层析分离得到式10所示吡喃酮104mg。(注:式10所示吡喃酮需先乙酰化才进行下一步还原反应)
取式10所示吡喃酮79mg,加入8mldcm,再加入乙酸酐16.5mg,呲啶12.7mg,dmap2.1mg,在室温下反应3小时,反应结束后使用氯化铵水溶液淬灭反应,萃取浓缩得到粗产物。
将上述粗产物56mg置于烧瓶中,在0℃下加入甲醇2ml,4.5mg硼氢化钠,在室温下反应1小时,反应结束后使用氯化铵水溶液淬灭反应,使用dcm萃取并浓缩得到烯醇粗产物,将烯醇粗产物溶解于2mldcm中,冷却至-78℃,加入2,6-二甲基吡啶0.06ml,三氟甲磺酸酸酐0.05ml,在-78℃下反应4小时,反应结束后使用氯化铵水溶液淬灭反应,使用dcm萃取并浓缩得到闭环粗产物。
最后将闭环粗产物溶于2ml甲醇中,并加入碳酸钾30mg,反应结束后先把溶剂浓缩,再溶于dcm中,使用氯化铵水溶液和1m盐酸淬灭反应,使用dcm萃取并浓缩得到粗产物,再一步把粗产物加到2ml的thf中,再加入1m四正丁基氟化铵的thf溶液0.12ml,在室温下反应半小时,反应结束后把溶剂浓缩,通过柱层析分离得到式1所示化合物24mg。该式1所示的化合物进行核磁检测,得到的1h-nmr图谱如图1所示,13c-nmr图谱如图2所示。
(2)一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子,分子结构如式2所示,其制备方法包括以下步骤:
只需用式8所示重氮盐取代式7所示重氮盐,得到式11所示的吡喃酮;其他步骤与上述式1所示的化合物制备流程相似,最终得到式2所示的化合物。将式2所示的化合物进行核磁检测,得到的1h-nmr图谱如图3所示,13c-nmr图谱如图4所示。
(3)一种以甲型流感病毒rna聚合酶为靶点的抗病毒药物musellarin及类似物分子,分子结构如式2所示,其制备方法包括以下步骤:
只需用式9所示重氮盐取代式7所示重氮盐,得到式12所示的吡喃酮;其他步骤与上述式1所示的化合物制备流程相似,最终得到式3所示的化合物。将式3所示的化合物进行核磁检测,得到的1h-nmr图谱如图5所示,13c-nmr图谱如图6所示。
实施例2半致死浓度(lc50)实验
准备所需培养介质和溶液:准备mirus公司
细胞培养:在t-75cm2的烧瓶中培养hek-293t的细胞,直到细胞密度达到70%-80%,并连续传代3次。实验前24小时收集细胞,每个96孔板接种约为2×106个细胞,并孵育细胞过夜。
生成转染复合物:准备1.43ml不含血清的介质,转染并加入22μg的dna质粒,加入66μl的transit-1l,混合均匀。在室温下孵化15-30分钟使形成转染复合物,在96孔板上每个孔中加入13μl的预混液。
将标准培养基中的细胞胰蛋白酶化,加入适量培养基,混合均匀,使用血球计数器确定每毫升培养介质的细胞数量。确保每孔中的细胞数量每毫升不少于2-4×104。在96孔板的每个孔中加入87μl稀释的细胞溶液。
将正常细胞转染甲型流感病毒聚合酶的质粒,然后将转染的细胞分别与式1所示化合物、式2所示化合物、式3所示化合物以及t-705相互接触。将药物分子加入到细胞中培养,测量药物分子对细胞生长的影响。等待40小时后收获细胞。进行乳酸脱氢酶实验,定量得到对应的半致死量浓度。
由于乳酸脱氢酶(lactatedehydrogenase)只存在于细胞内部,如果有机小分子杀死细胞,乳酸脱氢酶会被释放到溶液中,通过测量在溶液中乳酸脱氢酶的数量,进而可定量测得有机分子对细胞的毒性。在40小时的慢性试验中,由乳酸脱氢酶试验测得,如图8所示,将数值拟合到希尔方程曲线(hill–langmuirequation)计算lc50:式1所示化合物、式2所示化合物、式3所示化合物及t-705的毒性数值分别为:2273.95μm,2648.70μm,2511.85μm,3359.64μm。
由该实验可知:本发明实施例提供的化合物药物分子不具有诱导细胞溶解的能力,表现为较低的细胞毒性,表明该化合物的分子安全性好。以商品抗流感药物t-705作为参考,本发明实施例提供的化合物显示较低的毒性。
实施例3半抑制浓度(ic50)实验
分别准备浓度梯度为1000μm,200μm,40μm,8μm,1.6μm的式1所示化合物、式2所示化合物、式3所示化合物的药物分子溶液。
将实施例2中正常细胞转染流感病毒聚合酶dna,并分别加入药物分子溶液形成共转染的流感聚合酶质粒系统。细胞与药物分子在所准备的浓度梯度溶液下相互接触培养40小时后,由荧光素酶实验(luciferaseassay)测得半抑制浓度ic50。如图9所示,将数值拟合到希尔方程曲线(hill–langmuirequation)计算ic50):式1所示化合物、式2所示化合物、式3所示化合物、t-705的ic50分别为:12.54μm、28.48μm、6.3μm、19.05μm。
从实验结果可以看出:抗病毒化合物均具有抑制流感病毒聚合酶复合体活性的作用。本实施例证明,式1所示化合物、式2所示化合物、式3所示化合物能够抑制流感病毒的复合酶活性。
实施例4抑制流感病毒聚合酶体外转录的实验
准备纯化的流感病毒聚合酶、放射性同位素标记的rna引物、rna模板、ntp底物的体外转录混合物溶液。将1000μm,100μm,10μm,1μm,0.1μm,0.01μm浓度的式1所示的化合物和t-705-rtp加入上述体外转录混合物溶液中,在30℃孵育60分钟。
通过电泳凝胶将转录产物分离,根据放射性同位素在凝胶中的相对位置及强度测量不同浓度的式1所示的化合物及t-705-rtp(t-705-rtp是将t-705作为碱基直接连接在核苷三磷酸上,在体外实验中不需经细胞转化就可以直接参加酶聚合反应)对流感病毒聚合酶的直接抑制作用。如图10所示,将数值拟合到希尔方程曲线(hill–langmuirequation)计算icinvitro50:式1所示的化合物和t-705-rtp的icinvitro50分别为:0.15μm、29.56μm。
由该实验可知:式1所示的化合物能够抑制流感病毒的复合酶活性。
实施例5药物的选择性系数(selectivityindex)实验
选取法匹拉韦的有效成分t-705作为参考,根据实施例2和实施例3,通过公式:选择性系数=lc50/ic50,计算式1所示化合物、式2所示化合物、式3所示化合物的选择性系数,分别为181.34、93.02、399.02,显着优于t-705的选择性系数176.33。
其中式1所示化合物、式3所示化合物具有比t-705具有更优的药物选择性,表现为更好的选择性,可有效抑制流感病毒的rna聚合酶而不损伤正常的细胞,可针可针对该抑制剂分子进行分子设计以制备抗流感药物。
实施例6药物分子结合靶点的分子模拟分析
采用分子对接(moleculardocking)方法研究了式1所示化合物、式2所示化合物、式3所示化合物与甲型流感病毒的rna聚合酶结合的位点和结合的能量。式1所示化合物、式2所示化合物、式3所示化合物与流感病毒rna聚合酶结合位置示意图见图7,式1、式2、式3结构相似,可表现为与rna聚合酶相似的结合位点。
通过分子对接的结果发现:式1所示化合物、式2所示化合物、式3所示化合物可有效结合在两个位置。其一,式1、式2、式3所示的化合物可能结合在rna聚合酶的底物进入信道的位置,其计算得到的结合自由能分别为-8.9kcal/mol,-8.8kcal/mol,-9.1kcal/mol,其数值均接近磷酸化的t-705的结合自由能-9.0kcal/mol。式1、式2、式3所示的化合物可通过与leu642和asn671相互作用,有效结合在rna聚合酶的信道位置,通过阻断rna聚合酶的底物进入进而抑制rna聚合酶的转录和复制过程。其二,式1、式2所示的化合物可结合在甲型流感病毒pb2链的rna帽结构结合域中,计算所得的结合自由能为-8.7kcal/mol,-8.8kcal/mol。如图7所示,式1、式2可结合在rna聚合酶rna帽结构结合域中,与asn429、tyr432、arg332形成有利的结合,阻挡了rna聚合酶与其底物的结合。
该分子对接的结果显示:式1、式2、式3所示的化合物可以有效结合到rna聚合酶的内部,阻断底物进入通道即可有效抑制rna的转录和复制过程。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除
 热门咨询
热门咨询



