
 商标分类
商标分类  商标转让
商标转让 
一种阿替美唑的制备方法与流程
 2021-02-02 21:02:46|
2021-02-02 21:02:46| 511|
511| 起点商标网
起点商标网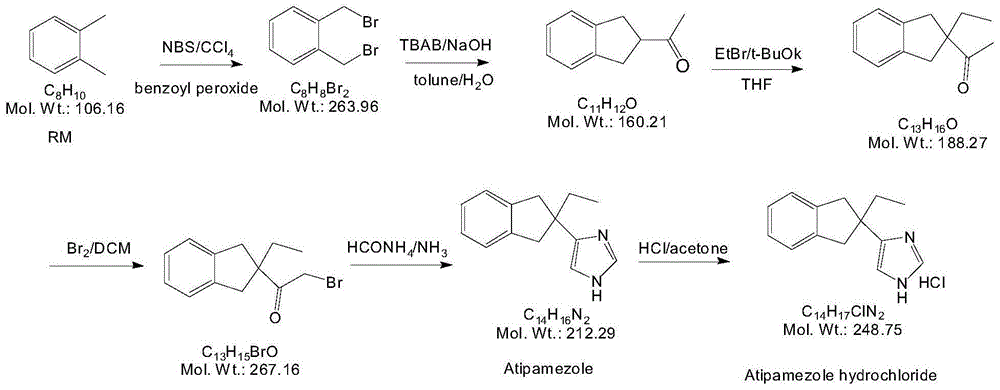
本发明属于兽用药制备方法技术领域,具体涉及一种阿替美唑的制备方法。
背景技术:
盐酸阿替美唑,化学名:4-(2-乙基-2-茚满)咪唑盐酸盐,商品名:antisedan,由美国orion-farmos公司开发。盐酸阿替美唑是一种合成的α2肾上腺素能受体拮抗剂,用于逆转犬使用右美托咪定和美托咪定后的镇静和镇痛作用。它的逆转作用通过与α2-肾上腺素能受体的镇静剂竞争性结合。阿替美唑对α2-肾上腺素能受体的高度特异性。副作用发生率低。阿替美唑主要用于兽医学,现许可用于狗和猫肌内静脉注射。其特点是:起效非常快,通常3-5分钟即可起效,10分钟左右达到最大血糖浓度,而其在血液中的半衰期小于3小时,随后通过在肝脏内进行生物转化,经尿液排出。由此可见盐酸阿替美唑是一种有效快捷安全的药物。另外,盐酸阿替美唑用于人类药物的研究也正在进行中。例如,盐酸阿替美唑用作降糖和降压方面的药物研究目前正处于临床评价研究阶段。有研究报道,盐酸阿替美唑在治疗哮喘、肥胖、偏头痛以及由年龄增长引起的记忆力衰退等方面都有一定的疗效。由于盐酸阿替美唑具有较大的药用应用前景,因此,开发盐酸阿替美唑的合成工艺具有重要的实用价值。
自美国orion-farmos公司开发首个盐酸阿替美唑合成工艺以来,国内外又研发了一些盐酸阿替美唑合成工艺,这些合成工艺各有特点。现总结如下。
(1)路线一如图1所示。该合成路线分为以下几步反应,第一步反应以邻二甲苯溴化成邻二溴甲苯,第二步再和乙酰丙酮在碱性条件及相转移催化剂存在下,经过中间产物2,2-二乙酰基茚满,合成了2-乙酰基茚满。第三步反应2-乙酰基茚满在无水四氢吠喃中,在叔丁基钾作用下与溴乙烷反应生成2-乙基-2-乙酰基茚满。
第四步反应2-乙基-2-乙酰基茚满与溴素反应生成2-(2-溴乙酰基)-2-乙基-茚满。第五步反应2-(2-溴乙酰基)-2-乙基-茚满于160℃与甲酰胺反应生成阿替美唑,再经盐酸酸化得到目标产物盐酸阿替美唑。
(2)路线二如图2所示。该合成路线分为四步反应,第一步反应以邻二溴甲苯和甲基烯丙基酮的钠盐为起始原料合成了2-乙酰基-2-乙烯基-茚满,第二步反应2-乙酰基-2-乙烯基-茚满在二氯甲烷中,常温下与液溴反应生成2-(2-溴乙酰基)-2-乙烯基-茚满,第三步反应2-(2-溴乙酰基)-2-乙烯基-茚满于160℃至180℃下与甲酰胺反应生成4-(2-乙烯基-2-茚满)咪唑,第四步反应4-(2-乙烯基-2-茚满)咪唑高压下在稀盐酸溶液中,钯碳催化下氢化反应生阿替美唑,再经盐酸酸化得到目标产物盐酸阿替美唑。
(3)路线三如图3所示。该合成路线分为五步反应,第一步反应以2-乙酰基茚酮和溴乙烷为起始原料,在无水碳酸钾和存在下反应,生成2-乙酰基-2-乙基茚酮,第二步反应2-乙酰基-2-乙基茚酮于二氯甲烷中经液溴溴化,常温下制备2-(2-溴乙酰基)-2-乙基茚酮,第三步反应2-(2-溴乙酰基)-2-乙基茚酮于160℃下与甲酰胺反应生成4-(2-乙基-2-茚酮)咪唑,第四步反应4-(2-乙基-2-茚酮)咪唑经硼氢化钠还原生成4-(2-乙基-2-茚醇)咪唑,第五步反应4-(2-乙基-2-茚醇)咪唑在稀盐酸溶液中,钯碳催化下氢化反应生成阿替美唑。
(4)路线四如图4所示。
(5)路线五如图5所示。该路线以三苯甲基保护的咪唑碘代物为起始原料,依次经过转变成醛,再与苯酞缩合,乙酰化,酸性脱去保护基,再氢化成阿替美唑。
上述工艺路线均存在不同程度问题。第一条工艺合成路线,尽管起始原料易得,价格相对较便宜,但是存在突出问题,例如,中间产物邻二溴苯刺激性极大;环合反应副反应多,精制困难。
对于第二条合成路线,虽然也比较适合工业化的生产,但其起始原料甲基烯丙基酮国内尚无稳定的供应,这一点限制了该路线的应用。
对于第三条路线,其路线也较为简洁,反应条件温和,易于实现工业化操作。但该路线起始原料2-乙酰基茚酮供应商很少,我们可以考虑用茚酮和咪唑自己合成,需要两步反应。
第四条路线采用亚硝基甲烷和氯气以及水合肼还原,反应条件严苛,有一定的危险性。并且对反应设备要求高。
第五条工艺路线最适合工业化生产,但文献报道的最后一步还原,同样用到盐酸做溶剂的催化氢化反应,常规高压釜都是不锈钢材质,无法耐受盐酸参与的高压反应。急需一种更适合工业放大方法来生产盐酸阿替美唑。
技术实现要素:
本发明要解决的技术问题是提供一种阿替美唑的制备方法,本制备方法的反应条件温和,重量收率可以达到74%以上,纯度为99%以上。
本发明的内容为一种阿替美唑的制备方法,步骤为,将化合物1经过两步还原,制备得到阿替美唑;反应路线如下:
在第二步中,将化合物2分散在有机溶剂中,加入还原剂三甲基碘硅烷,反应得到阿替美唑。
优选的,在第二步中,有机溶剂为乙腈和二氯甲烷的混合溶剂,反应温度为-20~-10℃。
在第一步中,将化合物1和还原剂在醇类溶剂中反应得到化合物2。所述还原剂优选为硼氢化钠、硼氢化钾、硼氢化锂中的一种或多种。醇类溶剂优选为乙醇、甲醇、异丙醇的一种或多种。反应温度为0-5℃。
本发明的有益效果是,本发明提供了一种制备阿替美唑的新路线,该路线反应条件温和,避免危险的高压反应,所得产品符合兽药典要求,重量收率可以达到74%以上,纯度99%以上。
本发明的化合物2在乙腈和二氯甲烷的溶剂中,被还原剂还原,重量收率可以达到92%以上。本还原剂选择性高,混合溶剂比单一溶剂,得到的产物收率高,纯度好。
本发明采用两步还原,而不是一步还原,避免了苛刻的反应条件,避免了杂质的生成,产物收率和纯度都非常好。
附图说明
图1为背景技术中路线一的反应路线图。
图2为背景技术中路线二的反应路线图。
图3为背景技术中路线三的反应路线图。
图4为背景技术中路线四的反应路线图。
图5为背景技术中路线五的反应路线图。
图6为本发明的反应路线图。
具体实施方式
本发明实施例中所用的各种化学品和试剂,均为市售购买。
实施例1制备化合物2
在0-5℃冷水浴中,取7g化合物1加入105ml无水乙醇(15v/w),搅拌,加入2.8g硼氢化钠,反应24小时,点板监控反应,待化合物1点消失,取丙酮30ml滴加到反应混合液中,终止反应。减压除去溶剂,加200ml二氯甲烷,搅拌2小时,过滤,减压除去溶剂,得化合物2粗品5.92克,收率:84.57%。
实施例2制备阿替美唑
取5.4g化合物2加入20ml乙腈中,再加入10ml二氯甲烷,降温至-20~-10℃,加入20ml三甲基碘硅烷反应,tlc检测反应完全,加入2m的硫代硫酸钠终止反应,静置分液,有机相用饱和食盐水洗涤一次,有机相浓缩去溶剂,加入2m盐酸50ml,在100℃反应1-1.5小时,过去保护基,过滤,水相用40%氢氧化钠调节ph>10,过滤得淡黄色固体2.3g。收率92%。
实施例3制备盐酸阿替美唑
取阿替美唑4克,在5℃冷水浴中,滴加两当量的盐酸乙醇(6m),滴加完搅拌30分钟;减压去除溶剂;再加入3-5(v/w)无水乙醇打浆3小时,温度25-30℃,过滤。滤饼在60℃减压烘干24h,得盐酸阿替美唑:3.81克,收率95.25%。
高效液相色谱检测:含量大于99%,单杂含量小于0.1%。
对比例1制备盐酸阿替美唑
取5.4g化合物2加入30ml乙腈中,降温至-20~-10℃,加入20ml三甲基碘硅烷反应,tlc检测反应产生3个杂质点,原料约剩余5%。加入2m的硫代硫酸钠终止反应,静置分液,有机相用饱和食盐水洗涤一次,有机相浓缩去溶剂,加入2m盐酸50ml,在100℃反应1-1.5小时,过去保护基,过滤,水相用40%氢氧化钠调节ph>10,过滤得淡黄色固体2.1g。收率84%。
取阿替美唑2.1克,在5℃冷水浴中,滴加两当量的盐酸乙醇(6m),滴加完搅拌30分钟;减压去除溶剂;再加入3-5(v/w)无水乙醇打浆3小时,温度25-30℃,过滤。滤饼在60℃减压烘干24h,得盐酸阿替美唑:1.89克,收率90%。高效液相色谱检测:纯度97%。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



