
 商标分类
商标分类  商标转让
商标转让 
一种合成纯化泰拉霉素杂质E的方法与流程
 2021-02-02 21:02:48|
2021-02-02 21:02:48| 377|
377| 起点商标网
起点商标网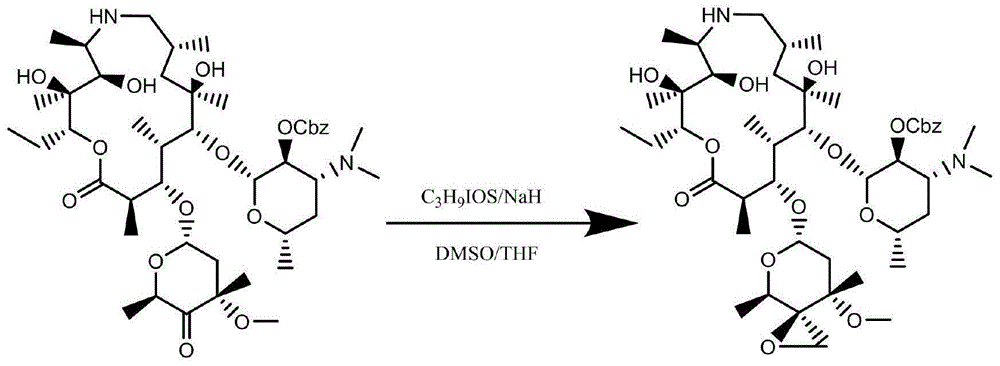
本发明属于药物化学技术领域,具体涉及一种合成纯化泰拉霉素杂质e的方法。
背景技术:
泰拉霉素(tulathromye)是一种动物专用的大环内酯类半合成抗生素,泰拉霉素主要用于放线杆菌、支原体、巴氏杆菌、副嗜血杆菌引起的猪、牛的呼吸系统疾病,具有用量少、一次给药、低残留和动物专用等众多优点。
目前,国内外泰拉霉素a的方法主要如下:以阿奇霉素a为原料,经cbz-cl得到保护的阿奇霉素a,然后经改进的pfitznor-moffat方法氧化,得到cbz保护的酮,酮通过wittig-horner反应,酮基转化为烯基得到保护的烯,保护的烯经氧化得到cbz保护的环氧化合物,cbz保护的环氧化合物在pd/c催化下脱保护得到脱保护的环氧化合物,脱保护的环氧化合物与正丙胺反应开环得到泰拉菌素粗品,粗品泰拉菌素与酸成盐纯化,然后解析得到泰拉菌素。
在车间生产泰拉霉素过程中,会出现多种杂质,杂质的出现对于研究泰拉霉素、泰拉霉素杂质的结构和性质方面起到了重要作用,但是通常出现的杂质纯度较低,无法满足对其结构和性质的研究条件,操作复杂,合成过程不易控制,单次制备产量较低,不能满足杂质质量控制需求,阻碍了对杂质的研究技术。
为了确保动物源性食品的安全,须对动物专用药物质量进行严格控制,药物含量测定、未知杂质的结构鉴定和杂质限量是药物质量控制的有效方法,杂质分析是药物质量标准的重要内容。泰拉霉素由半合成过程生产,与合成药物相比可控性更低,因此杂质谱更为复杂且难以预测。现行欧洲药典(ep)和美国药典(usp)均将兽药收录其中,并对有关物质限量提出要求,欧洲药典还要求对特定杂质进行控制(英国药典按惯例收录了欧洲药典的全部专论,内容一般不作修改)。vich(兽用药物注册技术要求国际协调会)指导原则要求兽医专用原料药的杂质报告限度为0.10%,鉴定限度为0.20%,控制限度为0.50%(该指导不包括半合成抗生素);ema(欧洲药品管理局)要求半合成兽用药物的杂质限度应符合vich指导原则的要求。
而泰拉霉素e为泰拉霉素合成过程中关键杂质,制备其标准品并对其结构进行研究都是泰拉霉素在国内申报兽药所必须的,同时对其结构进行研究也可以分析出它是在生产过程中何时产生,可以更好的减少其含量,从而更好的提高泰拉霉素纯度。
技术实现要素:
本发明提供一种合成纯化泰拉霉素杂质e的方法,解决现有技术中泰拉霉素杂质e的纯度低、操作复杂、合成过程不易控制、单次制备产量低、无法满足对其结构和性质的研究条件的问题。
具体技术方案如下:
一种合成纯化泰拉霉素杂质e的方法,包括以下步骤:
(1)向烧瓶加入碱溶液,溶剂,搅拌溶解,分次加入三甲基碘化亚砜,控制温度-10--20℃,该温度下保温搅拌2小时;滴加泰拉霉素氧化物的dmso溶液,控制滴加温度在-30--50℃,滴加完毕后于-50℃保温反应1小时,将反应液加入氯化铵水溶液中,淬灭反应,反应液分层,加盐水清洗后将有机相浓干,得环氧叉酮异构体粗品;
(2)环氧叉酮异构体粗品进行精制;
(3)脱保护过程:将精制后的环氧叉酮异构体在pd/c催化剂的作用下,脱去保护基;
(4)向步骤(3)中得到的脱保护基的环氧叉酮异构体,加入正丙胺、异丙醇,控制温度50-70℃,回流反应24小时;取样检测,若原料残留>10%,继续反应8-10小时,将反应液浓缩至干,得泰拉霉素杂质粗品e。
(5)泰拉霉素杂质e粗品精制。
优选的,所述步骤(1)中加入的碱溶液为叔丁醇钾、六甲基二硅基氨基钾或氢化纳中的一种;加入的溶剂为dmso和四氢呋喃的混合溶液;反应过程中通入n2。
优选的,环氧叉酮异构体粗品进行精制的方法为:将环氧叉酮异构体粗品加二氯甲烷溶解,滴加三氟乙酸和二氯甲烷混合溶液,继续滴加异丙醚,将析出的固体加水溶解,用碱溶液调节ph至8-9,并加入二氯甲烷萃取,二氯甲烷层浓干后得精制后的环氧叉酮异构体。
优选的,所述碱溶液为氢氧化钾或碳酸氢钾。
优选的,精制后的环氧叉酮异构体进行脱保护时,先用甲醇溶解,然后置于氢化釜中,加入pd/c催化剂,控制罐内氢气压力0.5mpa,30-35℃保温保压反应2小时,抽滤,滤液浓缩至干,得脱保护基的环氧叉酮异构体;
优选的,在脱保护过程中加入乙酸。
优选的,所述步骤(5)中对精制时的方法为:将泰拉霉素杂质e粗品与精制试剂混合后进行减压蒸馏,然后回流处理,降温后于0-5℃保温结晶,抽滤,烘干得到精制后的泰拉霉素杂质e。
优选的,将泰拉霉素杂质e粗品与精制试剂混合时,先向得到的泰拉霉素杂质e粗品中加入精制试剂,升温至全部溶解,再次加入精制试剂,升温至全部溶解。
优选的,所述精制试剂为二氯甲烷、正庚烷、乙酸乙酯、乙腈、石油醚。
有益效果:
1.本发明提供了一种合成纯化泰拉霉素杂质e的方法,最终得到的泰拉霉素杂质e性质稳定,纯度较高,操作简便,合成易于控制,能够满足行业内对于泰拉霉素、泰拉霉素杂质的结构和性质方面的研究要求。
2.本发明的合成纯化泰拉霉素杂质e的方法,调整了原料和反应温度,在dmso中,加入三甲基碘化亚砜,促使硫叶立德反应的进行,使其生成的环氧化合物以直立键为主,促进了泰拉霉素杂质e的转化效率,从而生成大量的泰拉霉素杂质e。
3.在进行脱保护过程中,加入乙酸到体系中,减少了二甲硫醚残留的影响,提高了pd/c活性,加快了反应效率,提高了泰拉霉素杂质e的纯度。
4.将泰拉霉素杂质e粗品与精制试剂混合时,先向得到的泰拉霉素杂质e粗品中加入精制试剂,升温至全部溶解,再次加入精制试剂,升温至全部溶解,使精制试剂与泰拉霉素杂质e充分混合,分子之间达到充分反应的要求,从而提高了泰拉霉素杂质e的质量以及泰拉霉素杂质e的纯度。
附图说明:
图1为环氧化过程示意图;
图2为脱保护基过程示意图;
图3为泰拉霉素杂质e生成过程示意图;
图4为泰拉霉素杂质e的cas号截图;
图5为泰拉霉素杂质e结构示意图;
图6为实施例1得到的泰拉霉素杂质e的核磁图谱;
图7为泰拉霉素对照品图谱;
图8为实施例1得到的泰拉霉素e对照品图谱;
ⅰ为泰拉霉素氧化物,ⅱ为环氧叉酮异构体,ⅲ为脱保护基的环氧叉酮异构体,ⅳ为泰拉霉素杂质e。
具体实施方式
图1为环氧化过程,反应式中(i)为泰拉霉素氧化物,在dmso以及四氢呋喃的混合溶液中,加入三甲基碘化亚砜和碱溶液,使其生成的环氧化合物以直立键为主,生成环氧叉酮异构体。
图2为脱保护基过程,反应式中(ii)为环氧叉酮异构体,它在pd/c的催化作用下,脱去保护基团,反应使用溶剂为甲醇,同时加入了适量的乙酸提高了pd/c活性。
图3为泰拉霉素杂质e生成过程,反应式中(ⅲ)为脱保护基的环氧叉酮异构体,该反应使用正丙胺对环氧化合物进行亲和加成反应,从而得到泰拉霉素e粗品,在经过分离、纯化得到泰拉霉素。
实施例1:
本实施例的合成纯化泰拉霉素杂质e的方法,包括以下步骤:
(1)向500ml四口烧瓶加入六甲基二硅基氨基钾3.0g,dmso和四氢呋喃共100ml,搅拌溶解,通入n2保护,分次加入三甲基碘化亚砜15.7g,控制温度0℃,该温度下保温搅拌2小时;滴加泰拉霉素氧化物的dmso溶液(30g+100ml),控制滴加温度在30℃,滴加完毕后于-50℃保温反应1小时,取样检测,环氧叉酮异构体:环氧化合物=85:15,将反应液加入10%氯化铵水溶液中,淬灭反应,反应液分层,加盐水清洗后将有机相浓干,得环氧叉酮异构体粗品30.4g;
(2)环氧叉酮异构体粗品进行精制:将环氧叉酮异构体粗品加二氯甲烷溶解,滴加三氟乙酸和二氯甲烷混合溶液,继续滴加异丙醚400ml,将析出的固体加水溶解,用碳酸氢钾调节ph至8,并加入二氯甲烷萃取,二氯甲烷层浓干后得精制后的环氧叉酮异构体;
(3)脱保护过程:将精制后的环氧叉酮异构体24.7g进行脱保护时,先用200ml甲醇溶解,然后置于氢化釜中,加入10%pd/c催化剂,控制罐内氢气压力0.5mpa,32℃保温保压反应2小时,抽滤,滤液浓缩至干,得脱保护基的环氧叉酮异构体23.3g,收率94.3%;在脱保护过程中加入乙酸;
(4)向步骤(3)中得到的脱保护基的环氧叉酮异构体23.3g,加入120mi正丙胺、120ml异丙醇,控制温度60℃,回流反应24小时;取样检测,原料残留6.3%,继续反应8小时,将反应液浓缩至干,得泰拉霉素杂质粗品e。
(5)泰拉霉素杂质e粗品精制:先向得到的泰拉霉素杂质e粗品中加入二氯甲烷与乙腈,升温至35℃全部溶解,再次加入精制试剂二氯甲烷与石油醚,升温至全部溶解,然后回流处理1h,降温后于1℃保温结晶2h,抽滤,烘干得到精制后的泰拉霉素杂质e6.2g,检测纯度,泰拉霉素e的纯度为91.67%,泰拉霉素的纯度为5.7%。
实施例2:
本实施例的合成纯化泰拉霉素杂质e的方法,包括以下步骤:
(1)向500ml四口烧瓶加入氢化纳3.0g,dmso和四氢呋喃共100ml,搅拌溶解,通入n2保护,分次加入三甲基碘化亚砜15.7g,控制温度20℃,该温度下保温搅拌2小时;滴加泰拉霉素氧化物的dmso溶液(30g+100ml),控制滴加温度在-30℃,滴加完毕后于-50℃保温反应1小时,取样检测,环氧叉酮异构体:环氧化合物=90:10,将反应液加入10%氯化铵水溶液中,淬灭反应,反应液分层,加盐水清洗后将有机相浓干,得环氧叉酮异构体粗品31.4g;
(2)环氧叉酮异构体粗品进行精制:将环氧叉酮异构体粗品加二氯甲烷溶解,滴加三氟乙酸和二氯甲烷混合溶液,继续滴加异丙醚400ml,将析出的固体加水溶解,用氢氧化钾调节ph至9,并加入二氯甲烷萃取,二氯甲烷层浓干后得精制后的环氧叉酮异构体;
(3)脱保护过程:将精制后的环氧叉酮异构体25g进行脱保护时,先用200ml甲醇溶解,然后置于氢化釜中,加入10%pd/c催化剂,控制罐内氢气压力0.5mpa,35℃保温保压反应2小时,抽滤,滤液浓缩至干,得脱保护基的环氧叉酮异构体24.2g,收率96.8%;在脱保护过程中加入乙酸;
(4)向步骤(3)中得到的脱保护基的环氧叉酮异构体24.2g,加入120mi正丙胺、120ml异丙醇,控制温度60℃,回流反应24小时;取样检测,原料残留7.9%,继续反应10小时,将反应液浓缩至干,得泰拉霉素杂质粗品e。
(5)泰拉霉素杂质e粗品精制:先向得到的泰拉霉素杂质e粗品中加入正庚烷与乙酸,升温至30℃全部溶解,再次加入精制试剂乙酸乙酯与正庚烷,升温至全部溶解,然后回流处理1h,降温后于3℃保温结晶2h,抽滤,烘干得到精制后的泰拉霉素杂质e5.6g,检测纯度,泰拉霉素e的纯度为93.4%,泰拉霉素的纯度为4.2%。
实施例3:
本实施例的合成纯化泰拉霉素杂质e的方法,包括以下步骤:
(1)向500ml四口烧瓶加入氢化纳3.0g,dmso和四氢呋喃共100ml,搅拌溶解,通入n2保护,分次加入三甲基碘化亚砜15.7g,控制温度10℃,该温度下保温搅拌2小时;滴加泰拉霉素氧化物的dmso溶液(30g+100ml),控制滴加温度在0℃,滴加完毕后于-50℃保温反应1小时,取样检测,环氧叉酮异构体:环氧化合物=90:10,将反应液加入10%氯化铵水溶液中,淬灭反应,反应液分层,加盐水清洗后将有机相浓干,得环氧叉酮异构体粗品30.0g;
(2)环氧叉酮异构体粗品进行精制:将环氧叉酮异构体粗品加二氯甲烷溶解,滴加三氟乙酸和二氯甲烷混合溶液,继续滴加异丙醚400ml,将析出的固体加水溶解,用碳酸氢钾调节ph至9,并加入二氯甲烷萃取,二氯甲烷层浓干后得精制后的环氧叉酮异构体;
(3)脱保护过程:将精制后的环氧叉酮异构体24.3g进行脱保护时,先用200ml甲醇溶解,然后置于氢化釜中,加入10%pd/c催化剂,控制罐内氢气压力0.5mpa,30℃保温保压反应2小时,抽滤,滤液浓缩至干,得脱保护基的环氧叉酮异构体23.8g,收率97.9%;在脱保护过程中加入乙酸;
(4)向步骤(3)中得到的脱保护基的环氧叉酮异构体23.8g,加入120mi正丙胺、120ml异丙醇,控制温度70℃,回流反应24小时;取样检测,原料残留6.8%,继续反应9小时,将反应液浓缩至干,得泰拉霉素杂质粗品e。
(5)泰拉霉素杂质e粗品精制:先向得到的泰拉霉素杂质e粗品中加入石油醚与乙酸乙酯,升温至40℃全部溶解,再次加入精制试剂正庚烷、二氯甲烷、乙酸乙酯,升温至全部溶解,然后回流处理1h,降温后于5℃保温结晶2h,抽滤,烘干得到精制后的泰拉霉素杂质e6.1g,检测纯度,泰拉霉素e的纯度为93.5%,泰拉霉素的纯度为4.7%。
实施例4:
本实施例的合成纯化泰拉霉素杂质e的方法,包括以下步骤:
(1)向500ml四口烧瓶加入叔丁醇钾3.0g,dmso和四氢呋喃共100ml,搅拌溶解,通入n2保护,分次加入三甲基碘化亚砜15.7g,控制温度-10℃,该温度下保温搅拌2小时;滴加泰拉霉素氧化物的dmso溶液(30g+100ml),控制滴加温度在50℃,滴加完毕后于-50℃保温反应1小时,取样检测,环氧叉酮异构体:环氧化合物=80:20,将反应液加入10%氯化铵水溶液中,淬灭反应,反应液分层,加盐水清洗后将有机相浓干,得环氧叉酮异构体粗品29.8g;
(2)环氧叉酮异构体粗品进行精制:将环氧叉酮异构体粗品加二氯甲烷溶解,滴加三氟乙酸和二氯甲烷混合溶液,继续滴加异丙醚400ml,将析出的固体加水溶解,用氢氧化钾调节ph至8,并加入二氯甲烷萃取,二氯甲烷层浓干后得精制后的环氧叉酮异构体;
(3)脱保护过程:将精制后的环氧叉酮异构体23.0g进行脱保护时,先用200ml甲醇溶解,然后置于氢化釜中,加入10%pd/c催化剂,控制罐内氢气压力0.5mpa,30℃保温保压反应2小时,抽滤,滤液浓缩至干,得脱保护基的环氧叉酮异构体21.5g,收率93.5%;在脱保护过程中加入乙酸;
(4)向步骤(3)中得到的脱保护基的环氧叉酮异构体21.5g,加入120mi正丙胺、120ml异丙醇,控制温度50℃,回流反应24小时;取样检测,原料残留7.2%,继续反应9小时,将反应液浓缩至干,得泰拉霉素杂质粗品e。
(5)泰拉霉素杂质e粗品精制:先向得到的泰拉霉素杂质e粗品中加入二氯甲烷与正庚烷,升温至30℃全部溶解,再次加入精制试剂二氯甲烷与乙腈,升温至全部溶解,然后回流处理1h,降温后于0℃保温结晶2h,抽滤,烘干得到精制后的泰拉霉素杂质e5.4g,检测纯度,泰拉霉素杂质e的纯度为92.8%,泰拉霉素的纯度为3.7%。
对比实验:
设置5组对比实验,各组对比实验的方法步骤同实施例1,不同之处在于对比例1将步骤(1)中的三甲基碘化亚砜替换为c3h9brs;对比例2中在步骤(1)的反应过程中不通入n2,对比例3在对精制后的环氧叉酮异构体进行脱保护时,将其直接置于常规反应釜中,其他参数同实施例1;对比例4不同之处在于脱保护过程中不加入乙酸;对比例5的不同之处在于将泰拉霉素杂质e粗品与精制试剂混合时,将精制试剂一次性与泰拉霉素杂质e混合。反应结束后,测定泰拉霉素杂质e的质量、纯度以及泰拉霉素的纯度,结果如下表所示。
从上表中可以看出,相对于实施例1的合成纯化方法,对比例1-5中得到的泰拉霉素杂质e的质量、泰拉霉素杂质e的纯度、泰拉霉素的纯度均有所下降;对比例1将步骤(1)中的三甲基碘化亚砜替换为c3h9brs,三甲基碘化亚砜的叶立德反应受热力学控制,促进了环氧化合物的直立键的形成,改变了传统的生产过程中的动力学反应,更有利于后续反应的进行。对比例2在步骤(1)的反应过程中不通入n2,没有氮气的保护氛围,其他杂质分子极易与原料反应,从而影响了最终泰拉霉素杂质e的纯度,通入氮气对整个反应体系进行了保护,有利于反应的进行;对比例3在对精制后的环氧叉酮异构体进行脱保护时,将其直接置于常规反应釜中,可以看出,在没有氢气的环境下,泰拉霉素杂质e的质量、泰拉霉素杂质e的纯度、泰拉霉素的纯度均有所下降,这表明若置于常规反应釜中,导致环氧叉酮异构体进行脱保护无法顺利进行,其保护基团无法完全脱去,影响了后续泰拉霉素e的合成过程。对比例4不同之处在于脱保护过程中不加入乙酸,pd/c的活性降低,导致反应速率减慢,反应不完全;对比例5的不同之处在于将泰拉霉素杂质e粗品与精制试剂混合时,将精制试剂一次性与泰拉霉素杂质e混合,无法使精制试剂与泰拉霉素杂质e充分反应,分子之间的反应无法达到充分反应的要求,而本发明分次进行了两次混合,能够使得精制试剂与泰拉霉素杂质e反应充分,从而提高了泰拉霉素杂质e的质量、泰拉霉素杂质e的纯度、泰拉霉素的纯度。
泰拉霉素杂质e的分子式:c41h80n3o12。
图4为泰拉霉素杂质e的cas号截图。
图6为实施例1得到的泰拉霉素杂质e的核磁图谱;图7为泰拉霉素a对照品图谱;图8为实施例1得到的泰拉霉素e对照品图谱;从图7中可以看出,泰拉霉素a在该流动相条件下保留时间为22.279;而泰拉霉素e相对泰拉霉素保留时间为1.09左右,因此泰拉霉素e的保留时间为24.28左右,从图8可以,保留时间为24.103这个物质与泰拉霉素e保留时间吻合,其纯度为91.67%。
泰拉霉素e在cdcl3中的1h-nmr谱测定数据如下:
泰拉霉素e在cdcl3中的13c-nmr数据如下:
显然本发明具体实现并不受上述方式的限制,只要采用了本发明的方法构思和技术方案进行的各种非实质性的改进,或未经改进将本发明的构思和技术方案直接应用于其它场合的,均在本发明保护范围之内。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



