
 商标分类
商标分类  商标转让
商标转让 
一种神秘果叶多糖及其制备方法与应用与流程
 2021-02-02 21:02:15|
2021-02-02 21:02:15| 450|
450| 起点商标网
起点商标网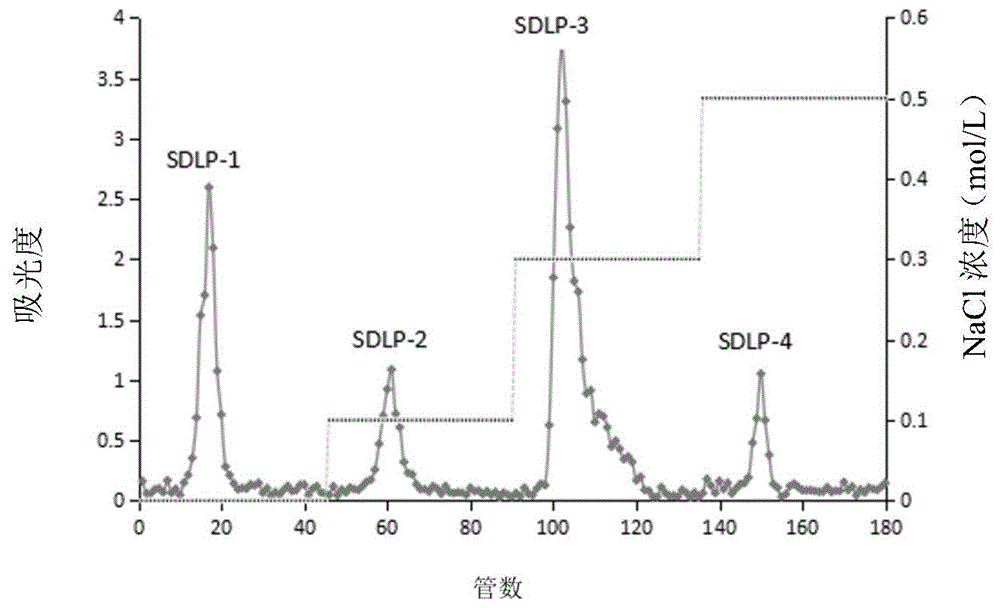
本发明属于多糖制备的技术领域,具体涉及一种神秘果叶多糖及其制备方法与应用。
背景技术:
神秘果(synsepalumdulcificum),是一种常绿的灌木植物,为山榄科神秘果属。原产于西非热带地区,目前在我国海南、云南等一些处于热带或亚热带地区都有种植。神秘果叶中的水溶性多糖的含量高达93.38mg/g。植物多糖具有多种生物活性,如抗氧化、抗衰老等。但由于神秘果种植环境要求高,其多糖的生产成本高,限制了它的开发和利用。因此以神秘果叶为研究对象,高效提取其中的多糖,对其在食品和医药行业的应用具有重要的意义。
不同植物多糖,具有不同的生物活性。而同一植物多糖,含有不同组分。现有的神秘果叶多糖采用乙醚、乙醇进行提取,不同溶剂下,提取的神秘果叶多糖成分不同,效果不同。而且现有技术中对于神秘果叶多糖进行分离纯化研究较少。
技术实现要素:
为了克服现有技术的缺点和不足,本发明的目的在于提供一种神秘果叶多糖及其制备方法。本发明将神秘果叶通过水煮沸提取,醇沉,脱蛋白处理、脱色处理,deae-52纤维素阴离子交换柱层析分离以及sephacryl-s200丙烯葡聚糖凝胶柱进一步分离提纯,获得五种多糖组分。
本发明的另一目的在于提供上述神秘果叶多糖的应用。所述神秘果叶多糖用于药物、食品和保健品领域,特别是抗氧化的药物、食品和保健品领域。
本发明的目的通过以下技术方案实现:
一种神秘果叶多糖的制备方法,包括如下步骤:
1)提取神秘果叶多糖:将晒干的神秘果叶粉碎,采用水对神秘果叶粉末进行煮沸提取,将提取液离心,取上清液,醇沉,冷冻干燥,获得神秘果叶粗多糖;所述醇沉是指向提取液中加入乙醇水溶液,在0~4℃静置24h以上;
2)脱蛋白处理:将神秘果叶粗多糖溶于水中,加入sevag试剂进行脱蛋白处理,获得脱蛋白的多糖溶液;所述sevag试剂为体积比为4:1的氯仿和正丁醇;
3)脱色处理:向脱蛋白的多糖溶液中加入氨水,调节ph至碱性,加入h2o2溶液,加热反应,离心,上清液减压浓缩,冷冻干燥,获得神秘果叶粗多糖;所述ph为8,所述加热反应的条件为55~65℃反应2~3h;
4)deae-52纤维素阴离子交换柱层析:将deae-52纤维素填料经预处理后装入层析柱,获得deae-52纤维素阴离子交换层析柱;将神秘果叶粗多糖溶于水,离心,取上清液,置于deae-52纤维素阴离子交换层析柱中,依次采用0、0.1、0.3、0.5mol/lnacl溶液进行洗脱,收集洗脱液;采用苯酚-硫酸法对洗脱液的多糖含量进行测定,绘制洗脱曲线,并以此为依据合并相同流分;减压浓缩,透析,冷冻干燥,得到多种多糖,分别记为多糖sdlp-1、sdlp-2、sdlp-3和sdlp-4;
5)sephacryl-s200丙烯葡聚糖凝胶柱分离纯化:将sephacryl-s200丙烯葡聚糖凝胶填料经预处理后装柱,获得预处理的凝胶柱;将多糖sdlp-1和sdlp-3分别溶于水,离心,取上清液,置于sephacryl-s200丙烯葡聚糖凝胶柱中,用水洗脱,收集洗脱液,采用苯酚-硫酸法对洗脱液的多糖含量进行测定,绘制洗脱曲线,并以此为依据合并相同流分;减压浓缩,冷冻干燥,得到多糖产物,多糖sdlp-1经凝胶柱水洗脱仍为一个组分,记为纯化后的多糖sdlp-1;多糖sdlp-3经凝胶柱水洗脱得到两个组分,记为sdlp-3a和sdlp-3b。
所述纯化后的多糖sdlp-1为均一组分,分子量(mw)为1481kda;多糖sdlp-3a和sdlp-3b都为均一组分,分子量(mw)分别为1286.3kda,44.3kda。
所述纯化后的多糖sdlp-1中甘露糖:葡萄糖:半乳糖:阿拉伯糖的摩尔比为0.29:26.95:2.01:1.6。
多糖sdlp-3a中鼠李糖:葡糖糖醛酸:半乳糖醛酸:葡萄糖:半乳糖:阿拉伯糖的摩尔比为1.54:1.28:2:7.16:4.73:5.47。
多糖sdlp-3b中鼠李糖:葡糖糖醛酸:半乳糖醛酸:葡萄糖:半乳糖:阿拉伯糖的摩尔比为1.82:1.84:5.14:2.01:7.16:5.29。
多糖sdlp-2和sdlp-4为非均一组分。
步骤1)中所述乙醇水溶液为体积分数为80%的乙醇水溶液。
所述提取的次数为1~3次;每次提取时,神秘果叶粉末与水的质量体积比为1g:(15~20)ml。提取的时间为1~5h。
步骤1)中所述离心的转速为4500~5500rpm,离心的时间为3~8min。
提取液在离心前,进行浓缩,浓缩至原体积的40~80%。
步骤3)中所述离心的转速为4500~5500rpm,离心的时间为3~8min。
步骤3)中所述双氧水为质量分数为30%的h2o2溶液,30%h2o2溶液的加入量满足其加入后溶液开始褪色为止。
步骤4)中deae-52纤维素填料的预处理,包括:将填料采用水浸泡后抽滤,0.5mol/l的naoh溶液浸泡,抽干并用水洗至中性,0.5mol/l的hcl溶液浸泡,抽干并用水洗至中性,最后0.5mol/l的naoh溶液浸泡,抽干,去离子水洗至中性。氢氧化钠溶液、盐酸各自浸泡的时间为0.5~1.5h。水浸泡的时间为8~14h。
步骤4)中神秘果叶粗多糖溶于水中神秘果叶粗多糖与水的质量体积比为(15~25)mg:1ml,优选为20mg/ml。
步骤4)中所述离心的转速为5500~6500rpm,离心的时间为10~15min。
步骤4)中透析是在透析袋(3500da)中透析48h,每6h更换一次蒸馏水。
步骤5)中所述预处理是指将填料在水中浸泡8~15h。
步骤5)中将多糖sdlp-1和sdlp-3分别溶于水,多糖sdlp-1和sdlp-3各自的浓度为10mg/ml。
步骤5)中离心的转速为5500~6500rpm/min,离心的时间为10~15min。
所述多糖用作清除dpph自由基、abts+自由基、超阴离子自由基和/或去除金属离子的抗氧化剂。纯化后的多糖sdlp-1用于清除超阴离子自由基的抗氧化物;sdlp-3a用于去除金属离子的抗氧化物。
本发明的有益效果:本发明提供了一种神秘果叶多糖提取和分离纯化方法,先将神秘果叶粉末用水进行提取,再对提取液进行脱蛋白脱色素处理,经deae-52纤维素阴离子交换柱和sephacryl-s200丙烯葡聚糖凝胶柱分离纯化,得到神秘果叶多糖sdlp-1、sdlp-2、sdlp-3a、sdlp-3b和sdlp-4。纯化后的多糖sdlp-1,多糖sdlp-3a和sdlp-3b都为均一组分,分子量(mw)分别为1481kda,1286.3kda,44.3kda。纯化后的多糖sdlp-1中甘露糖:葡萄糖:半乳糖:阿拉伯糖的摩尔比为0.29:26.95:2.01:1.6。多糖sdlp-3a中鼠李糖:葡糖糖醛酸:半乳糖醛酸:葡萄糖:半乳糖:阿拉伯糖的摩尔比为1.54:1.28:2:7.16:4.73:5.47。多糖sdlp-3b中鼠李糖:葡糖糖醛酸:半乳糖醛酸:葡萄糖:半乳糖:阿拉伯糖的摩尔比为1.82:1.84:5.14:2.01:7.16:5.29。多糖sdlp-2和sdlp-4为非均一组分。本发明的多糖包括均一组分多糖和非均一组分多糖,各多糖的结构不同。本发明的多糖具有较好的抗氧化效果。本发明的提取和分离纯化方法效率高,工艺流程简单,应用前景广泛。
附图说明
图1是实施例1中神秘果叶粗多糖经deae-52纤维素阴离子交换柱层析分离后得到4个组分的洗脱曲线;
图2是实施例1中多糖sdlp-1和sdlp-3经sephacryl-s200丙烯葡聚糖凝胶柱洗脱曲线;
图3是实施例2中纯化后的多糖sdlp-1、多糖sdlp-3a和sdlp-3b的紫外吸收图谱;
图4是纯化后的多糖sdlp-1的红外光谱图;
图5是多糖sdlp-3a的红外光谱图;
图6是多糖sdlp-3b的红外光谱图;
图7是实施例1中多糖(csdlp、纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b)对dpph自由基清除率的曲线图;csdlp对应脱蛋白脱色后的粗多糖;
图8是实施例1中多糖(csdlp、纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b)对abts自由基清除率的曲线图;csdlp对应脱蛋白脱色后的粗多糖;
图9为实施例1中多糖(csdlp、纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b)对超阴离子自由基清除率的曲线图;csdlp对应脱蛋白脱色后的粗多糖;
图10为实施例1中多糖(csdlp、纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b)对fe2+螯合力曲线图;csdlp对应脱蛋白脱色后的粗多糖。
具体实施方式
下面结合具体实施例对本发明作进一步详细的描述,但本发明的实施方式不限于此。
实施例1
一种从神秘果叶中提取和分离纯化多糖的方法,包括以下步骤:
(1)提取神秘果叶多糖:称取1000g经粉碎和过60目筛的神秘果叶粉末置于容器,加15000ml蒸馏水,于回流装置中煮沸提取多糖,提取的时间为3h,过滤,再向滤渣加入15000ml蒸馏水重复提取,合并2次的提取液,5000rpm离心5min,弃沉淀,取上清液;向上清液加入4倍体积的80%(v/v)乙醇溶液并不断搅拌,置于4℃冰箱中静置沉淀24h,5000rpm离心10min,取出沉淀冷冻干燥,得到神秘果叶粗多糖;
(2)对神秘果叶粗多糖进行脱蛋白处理:取神秘果叶粗多糖溶于蒸馏水中,将粗多糖溶液置于分液漏斗中,加入体积是神秘果叶粗多糖溶液体积的1/4的sevag试剂(氯仿和正丁醇按4:1的体积比混合配制),充分振荡60min,静置使其分层,打开分液漏斗将有机试剂和夹在多糖溶液和有机试剂中间的蛋白质层分离出来,如此重复5~6次,直到没有蛋白质层出现;
(3)对脱蛋白的神秘果叶多糖溶液进行脱色素处理:向脱蛋白后神秘果叶多糖溶液,加入氨水调节ph至8.0,再加入30%h2o2溶液,直到多糖溶液开始褪色;将溶液置于60℃水浴反应2h;然后5000rpm离心5min,取上清液减压浓缩,冷冻干燥,制得脱蛋白脱色素后的神秘果叶粗多糖(csdlp);
(4)deae-52纤维素阴离子交换柱层析分离神秘果叶多糖:
(4-1)填料预处理:用去离子水浸泡deae-52纤维素过夜溶胀,抽滤,加入0.5mol/l的naoh溶液浸泡1h,抽干后用去离子水反复洗涤至中性;再加入0.5mol/l的hcl溶液浸泡1h,抽干后再用去离子水反复洗涤至中性;再加入0.5mol/l的naoh溶液浸泡1h,抽干后用去离子水反复洗涤至中性,备用;
(4-2)装柱:采用湿法装柱,将溶胀清洗后的deae-52纤维素装入层析柱中,装柱过程避免有气泡和分层出现,然后用去离子水平衡;
(4-3)上样:称取200mgcsdlp多糖,溶于10ml蒸馏水中,于6000rpm离心10min,取上清液,上到deae-52纤维素柱上;
(4-4)梯度洗脱:依次用3个柱体积的蒸馏水、0.1、0.3、0.5mol/lnacl溶液进行梯度洗脱,流速为1.0ml/min,每10ml收集一管,采用苯酚-硫酸法对洗脱液的多糖含量进行测定,以收集的管数作为横坐标,以每管对应所测得的吸光度作为纵坐标,绘制神秘果叶多糖溶液的deae-52纤维素离子交换柱洗脱曲线,如图1所示,根据洗脱曲线将相同组分的多糖合并;
(4-5)透析:将收集的不同组分,减压浓缩,装入截留分子量为3500da透析袋中密封放入蒸馏水中透析48h除盐,每6h更换一次蒸馏水,然后冷冻干燥,分别得到白色的多糖组分sdlp-1、sdlp-2、sdlp-3和sdlp-4。
(5)sephacryl-s200丙烯葡聚糖凝胶柱进一步分离神秘果叶多糖:用去离子水浸泡sephacryl-s200丙烯葡聚糖凝胶,过夜溶胀,然后采用湿法装柱,用3-4个柱体积的去离子水冲洗柱子和平衡;称取20mg不同组分的多糖(sdlp-1和sdlp-3),溶于2ml蒸馏水中,于6000rpm/min离心10min,取上清液上到sephacryl-s200丙烯葡聚糖凝胶上;用3个柱体积蒸馏水洗脱,流速为0.5ml/min,每10ml收集一管,采用苯酚-硫酸法对洗脱液的多糖含量进行测定,以收集的管数作为横坐标,以每管对应所测得的吸光度作为纵坐标,绘制神秘果叶多糖溶液的sephacryl-s200丙烯葡聚糖凝胶柱洗脱曲线,如图2所示,根据洗脱曲线将相同组分的多糖合并;收集的不同组分,减压浓缩,冷冻干燥后,分别得到纯化后的多糖sdlp-1,多糖sdlp-3a和sdlp-3b。
步骤(4-4)中苯酚-硫酸法检测多糖含量具体操作如下:分别精确吸取已配好的葡萄糖标准溶液0.1、0.2、0.3、0.4、0.5、0.6、0.7、0.8ml于干净的试管中,补蒸馏水至2.0ml,然后加入1.0ml5%苯酚溶液,振荡使其充分混匀,再迅速加入5.0ml浓硫酸,充分混匀。将试管置于40℃水浴15min后,取出迅速冷却到室温,用uv-2700型分光光度计在490nm处测定反应后溶液的吸光度,以葡萄糖溶液浓度为横坐标,以不同浓度的葡萄糖溶液对应紫外分光光计得到的吸光度为纵坐标,绘制葡萄糖标准曲线;量取样品溶液2ml,按标准曲线操作方法,测定其吸光度,计算样品溶液中总多糖的含量。
图1是实施例1中神秘果叶粗多糖经deae-52纤维素阴离子交换柱层析分离后得到4个组分的洗脱曲线;
图2是实施例1中多糖sdlp-1和sdlp-3经sephacryl-s200丙烯葡聚糖凝胶柱洗脱曲线。
实施例2
对实施例1得到的神秘果叶多糖进行紫外光谱分析,具体操作如下:
精确称取各组分多糖样品(纯化后的多糖sdlp-1,多糖sdlp-3a和sdlp-3b),用蒸馏水将样品溶解并配制成浓度为1.0mg/ml的不同组分多糖样品溶液,在200~400nm波长范围内进行紫外全波长扫描。
神秘果叶纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b的紫外吸收图谱结果如图3所示,各组分在260nm和280nm处均没有明显吸收峰,表明纯化后的sdlp-1、sdlp-3a和sdlp-3b不含蛋白质和核酸杂质。
实施例3
对实施例1得到的神秘果叶多糖进行分子量分析,具体操作如下:
采用高效凝胶渗透色谱法(hpgpc)测定神秘果叶多糖的纯度和分子量。将各纯化的多糖组分溶于流动相中,配制成浓度为2.0mg/ml的供试品溶液,经0.22μm滤膜过滤后备用。色谱条件:色谱柱为tskgelgmpwxl(7.8mm×30cm,13μm),流速为0.6ml/min,流动相为0.7%na2so4(含0.02%的proclin300抑菌剂),进样量为100μl,柱温为35℃,检测器为多角度激光散射器。精确称取分子量不同的葡聚糖标准品溶于流动相,配制成浓度为2.0mg/ml的标准品。以保留时间为横坐标,以信号值为纵坐标,绘制标准曲线,并据此计算出多糖的分子量。
测得神秘果叶多糖纯化组分的分子量如下表1所示:
表1纯化后的多糖sdlp-1、多糖sdlp-3a和sdlp-3b的分子量
实施例4
对实施例1得到的神秘果叶多糖进行单糖组成分析,具体方法如下:
采用pmp柱前衍生化法测定多糖组分中单糖的组成,通过hplc进行神秘果叶多糖各纯化多糖组分中单糖的定性定量分析。
样品酸水解:分别精确称取5mg纯化后的多糖sdlp-1、多糖sdlp-3a和sdlp-3b,分别装入具塞水解管中,加入2ml4mol/l三氟乙酸溶液,并充入氮气封管,放置于110℃鼓风烘箱中水解2h。取出水解管冷却至室温,加入5ml纯甲醇,在50℃下减压浓缩并蒸干,重复上述操作3次,溶液蒸干后加入1.0ml蒸馏水溶解水解后的样品。
样品的pmp衍生化:取上述水解后样品溶液200μl,转入1.5ml离心管中,加入200μl0.3mol/lnaoh溶液,摇匀,再加入400μl0.5mol/lpmp甲醇溶液,摇匀,密封离心管,70℃水浴30min,然后取出冷却至室温,加入200μl0.3mol/lhcl溶液中和反应溶液;60℃旋蒸至干,加入1.0ml蒸馏水溶解,然后加入1.0ml氯仿混匀,置于分液漏斗中,静置,除去氯仿和pmp衍生剂,重复上述操作3次,最后取出水相溶液用0.45μm滤膜过滤,以作液相分析备用。
混合标准品的pmp衍生化:取甘露糖、鼠李糖、葡萄糖醛酸、半乳糖醛酸、葡萄糖、半乳糖、阿拉伯糖和岩藻糖标准单糖混合,配制成10mmol/l的混标溶液,取200μl混标溶液按照上述的操作进行混合标准品的衍生化实验。
液相色谱条件:色谱柱:zorbaxeclipsexdb-c18(4.6mm×150mm,5μm);流动相:乙腈-pbs溶液(0.1mol/l,ph=6.7)=15:85;柱温:30℃;流速:1.0ml/min;检测波长:250nm;进样体积:10μl。
根据外标法以混合标准品中各单糖色谱峰的峰面积为纵坐标(y)和单糖所对应的浓度为横坐标(x)制作标准曲线;根据保留时间判断样品的单糖组成,根据各组成单糖的峰面积和标准曲线推算出多糖组分中组成单糖的摩尔比。
测得神秘果叶多糖的单糖组成结果如下表2:
表2纯化后的多糖sdlp-1、多糖sdlp-3a和sdlp-3b的单糖组成
实施例5
对实施例1得到的神秘果叶多糖进行红外光谱分析,具体方法如下:
分别取纯化后的多糖sdlp-1、多糖sdlp-3a和sdlp-3b各2mg,分别将样品放入atr红外光谱仪的进样盘中间,扫描范围为:4000-500cm-1,重复操作3次,通过傅里叶变换红外光谱仪得出红外谱图。
纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b的红外光谱图分别如图4-6所示。在3281.34、3279.95、3264.12cm-1处的吸收峰是由o-h伸缩振动产生,2928.86、2925.30、2931.31cm-1处的吸收峰是由c-h伸缩振动产生,1418.52和1419.35cm-1(sdlp-3a和sdlp-3b;sdlp-1此处无伸缩振动)处的吸收峰是由c-o伸缩振动产生,这些峰都是多糖的特征峰,可以判断纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b均属于多糖类物质。
纯化后的多糖sdlp-1的红外光谱图(图4)中,在1639.07cm-1处有明显的吸收峰,说明有水结合;1148.19、1077.55、1014.91cm-1处的吸收峰说明多糖中含有吡喃糖环的结构;933.07、761.49cm-1处的吸收峰说明多糖中存在d-吡喃葡萄糖;在(844±8)cm-1和(891±7)cm-1的吸收峰是吡喃糖环上α-和β-端基差异向异构体的c-h变角振动,因此在858.05cm-1处的吸收峰表明纯化后的多糖sdlp-1中存在α-糖苷键。
sdlp-3a的红外光谱图(图5)中,1020.07、1075.37、1143.80cm-1处的吸收峰说明多糖中含有吡喃糖环的结构;在890.91cm-1的吸收峰可以表明sdlp-3a中存在β-吡喃糖环型糖苷键;1720.99cm-1处有吸收峰,表明多糖sdlp-3a中含有糖醛酸。
sdlp-3b的红外光谱图(图6)中,1013.50、1047.26、1070.24cm-1的吸收峰说明多糖中含有吡喃糖环的结构;在1737.38cm-1处有明显的吸收峰,说明多糖sdlp-3b中含有糖醛酸。
图4是纯化后的多糖sdlp-1的红外光谱图;图5是多糖sdlp-3a的红外光谱图;图6是多糖sdlp-3b的红外光谱图。
实施例6
对实施例1得到的神秘果叶多糖进行抗氧化活性研究,具体方法如下:
多糖清除dpph自由基能力测定:分别精确吸取50μl不同浓度(0.3、0.5、1.0、2.0、3.0、4.0和5.0mg/ml)的神秘果叶多糖(csdlp、纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b)溶液和vc对照品溶液,加入96孔板中,再加入0.4mmol/ml的dpph溶液25μl和蒸馏水100μl,将96孔板中的溶液混合均匀充分反应,并置于阴暗处静置30min,使用酶标仪在517nm处测定吸光度,每个反应做三次平行重复实验,以vc组作为阳性对照。按照公式(6.1)计算dpph清除率。
其中:a0为蒸馏水替代样品做对照组实验吸光度;as1为样品实验吸光度;as2为无水乙醇替代dpph溶液做干扰组实验吸光度。
0.4mmol/ldpph溶液配制:精密称取15.7728mg的dpph粉末,溶于无水乙醇中,并定容至100ml。dpph清除率的测试结果如图7所示。
当浓度达到5mg/ml时,sdlp-1的dpph清除率为51.74%。
多糖清除abts+自由基能力测定:分别精确吸取50μl不同浓度(0.3-5.0mg/ml)的神秘果叶多糖(csdlp、纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b)溶液和vc对照品溶液,加入96孔板中,再加入200μl的abts+工作液,充分混合,置于室温下反应6min,于734nm波长下测定吸光度,每个反应做三次平行重复实验,以vc组作为阳性对照。按照公式(6.2)计算abts+自由基清除率。
其中:a0为蒸馏水替代样品溶液做对照组实验吸光度;as1为样品实验吸光度;as2为pbs替代abts+工作液做干扰组实验吸光度。
abts储备液:配制浓度为7mmol/l的abts溶液和4.95mmol/l的过硫酸钾溶液,按照体积比为1:1充分混合,于室温暗处下静置12h,备用。
abts+工作液配制:将abts储备液用0.2mol/lpbs(ph=7.4)溶液稀释至在734nm波长下的吸光度为0.70±0.02左右即可。
abts+清除率的测试结果如图8所示。
当浓度达到5mg/ml时,sdlp-1的abts+清除率为55.15%。
多糖清除超氧阴离子自由基能力测定:精确吸取50μl不同浓度(0.3-5.0mg/ml)的神秘果叶多糖(csdlp、纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b)溶液和vc对照品溶液,加入96孔板中,再各加入50μl156μmol/lnbt溶液、468μmol/lnadh溶液和60μmol/lpms溶液,充分混合,置于室温下反应5min,于560nm波长下测定吸光度,每个反应做三次平行重复实验,以vc组作为阳性对照。按照公式(6.3)计算超氧阴离子自由基清除率。
其中:a0为蒸馏水替代样品溶液做对照组实验吸光度;as1为样品实验吸光度;as2为0.1mol/l磷酸缓冲液(ph=7.4)替代nbt溶液做干扰组实验吸光度。
超氧阴离子自由基清除率的测试结果如图9所示。
当浓度达到5mg/ml时,sdlp-1的超氧阴离子清除率为94.39%。
多糖金属离子螯合能力测定:精确吸取50μl不同浓度(0.3-5.0mg/ml)的神秘果叶多糖(csdlp、纯化后的多糖sdlp-1、sdlp-3a和sdlp-3b)溶液和edta-2na对照品溶液,加入96孔板中,然后依次加入2.5μl2.0mmol/l的fecl2溶液、10μl5.0mmol/l菲啰嗪溶液和137μl的蒸馏水,充分混合,置于室温下反应10min,于562nm波长下测定吸光度,每个反应做三次平行重复实验,以edta-2na组作为阳性对照。按照公式(6.4)计算金属离子螯合能力。
其中:a0为蒸馏水替代样品溶液做对照组实验吸光度;as1为样品实验吸光度;as2为蒸馏水替代fecl2溶液做干扰组实验吸光度。
金属离子螯合能力测试结果如图10所示。
当浓度达到5mg/ml时,sdlp-1的金属离子清除率为84.17%。
图7是实施例1中多糖对dpph自由基清除率的曲线图;图8是实施例1中多糖对abts自由基清除率的曲线图;图9为实施例1中多糖对超阴离子自由基清除率的曲线图;图10为实施例1中多糖对fe2+螯合力曲线图。
对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下还可以做出若干简单推演或替换,而不必经过创造性的劳动。因此,本领域技术人员根据本发明的揭示,对本发明做出的简单改进都应该在本发明的保护范围之内。上述实施例为本发明的优选实施例,凡与本发明类似的工艺及所作的等效变化,均应属于本发明的保护范畴。
起点商标作为专业知识产权交易平台,可以帮助大家解决很多问题,如果大家想要了解更多知产交易信息请点击 【在线咨询】或添加微信 【19522093243】 与客服一对一沟通,为大家解决相关问题。
与客服一对一沟通,为大家解决相关问题。
此文章来源于网络,如有侵权,请联系删除

 热门咨询
热门咨询



